Block Copolymers
![]()
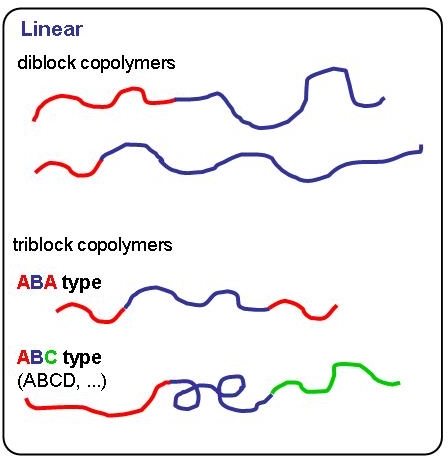
Block copolymers are normally prepared by controlled polymerization of one monomer, followed by chain extension with a different monomer(1) to form AB or ABC block copolymers. If you have not read the section on "Chain Extension" in the "Procedures for Initiation of an ATRP" you should return to that section if you wish to efficiently chain extend an existing macroinitiator.
When ATRP is employed to form each block in a block copolymer the composition and molecular weight and, to some extent, the PDI of each block can be independently controlled. Multifunctional initiators can also be used in the process to prepare ABA or AB-star multi-armed block copolymers.(2,3)
Macroinitiators can be prepared by any polymerization process including free radical polymerization(4) and other controlled polymerization processes including cationic,(5,6) anionic,(7) ROP,(8) condensation(9) and post-metallocene catalysis(10) as long as the terminal functionality is, or can be, converted to an ATRP initiating moiety. Any type of ATRP catalyst/initiation system can be employed.(11)
Targeted applications range from thermoplastic elastomers (12) to precursors for carbon nanostructures,(13,14) indeed one material can meet the requirements of both applications.(15) With the development of CRP it has been determined that the properties of the materials targeting thermoplastic elastomer applications can be significantly affected by the mode of (co)polymerization of the second block.(12) In a continuous ATRP copolymerization the fraction of first comonomer available for incorporation into the second block significantly changes the properties of the final material by preparation of a gradient copolymer segment in the second block.(16, 17) The formation of "gradient" copolymer A-blocks affects phase separation.
The term "normally" was used in the first sentence to allow for the fact that another method for preparation of multi-segmented block copolymers has been developed based on "click" chemistry.(18) α,ω-Diazido-terminated polystyrene-b-poly(ethylene oxide)-b-polystyrene was coupled with propargyl ether in DMF in the presence of a CuBr/N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA) catalyst. The same catalyst could be used for both the formation of the first precursor block copolymer and the chain extended multiblock copolymer as shown below.(19,20)
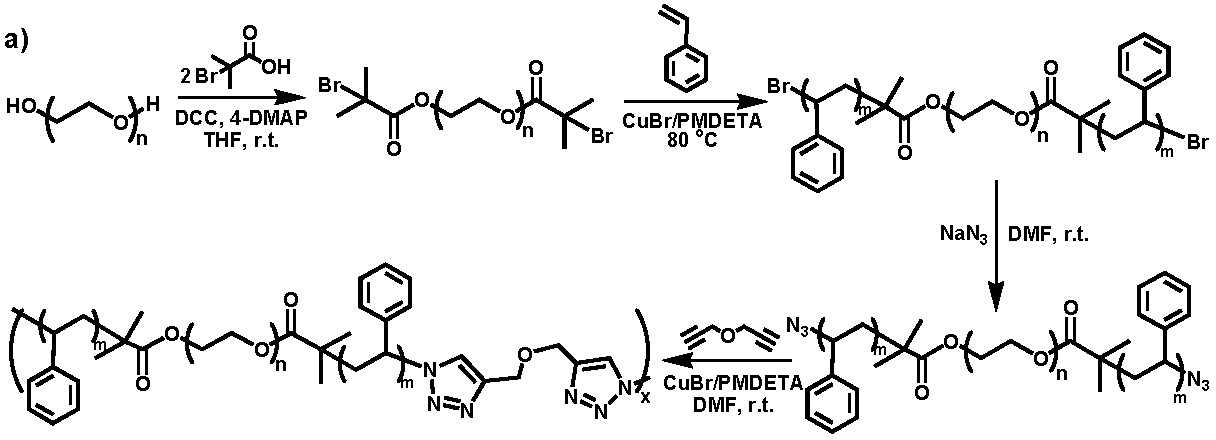
The strategy involves one or two chain extensions in a normal ATRP block copolymerization followed by end group transformation(19) and click coupling to obtain copolymers with up to 25 polymer segments in a single chain.(20) Formation of a product of higher molecular weight and broader molecular weight distribution was verified by triple-detection size exclusion chromatography, which revealed that typically 5-7 ABA block copolymers were linked together during the click chain extension reaction. Differential scanning calorimetry and dynamic mechanical analysis revealed that the amphiphilic block copolymer behaves as a viscoelastic fluid, while its corresponding multiblock copolymer is an elastic material. This would suggest that the polystyrene domains aggregate to form chemical cross-links in the swellable gel
A paper by Lutz(21) describes the use of "click" ligation of an amphiphilic block copolymer (AB type) and a hydrophilic homopolymer (B type) to prepare amphiphilic block copolymers. Well-defined ω-azido functional diblock copolymer PS-b-POEGA and α-alkyne functional homopolymer POEGA were prepared using ATRP and the two polymers could be efficiently coupled to each other via copper-catalyzed azide-alkyne click chemistry in aqueous medium. Moreover, using this coupling strategy, an ester group was introduced at the junction between AB and B segments. This labile moiety may be "cut" by hydrolysis providing a degradable system.
Copper-catalyzed azide-alkyne "click" chemistry was also applied to post-polymerization crosslinking of block copolymer formed by RAFT to control copolymer structure and the ability of the modified polymeric materials for subsequent self-assembly in response to changes in temperature.(22)
It is now possible to extend the range of monomers incorporated into a block copolymer by conducting successive RAFT polymerization and ATRP employing a dual functional bromoxanthate iniferter (initiator-transfer agent-terminator).(23) Poly(vinyl acetate)-b-polystyrene, poly(vinyl acetate)-b-poly(Me acrylate) and poly(vinyl acetate)-b-poly(Me methacrylate) block copolymers with low polydispersity (Mw/Mn < 1.25) were prepared by successive RAFT polymerization and ATRP employing a bromoxanthate iniferter (initiator-transfer agent-terminator) and a single catalyst complex for both steps.
A further advantage of this low catalyst transition metal mediated group transfer agent polymerization process is a further reduction inside reactions resulting in an ability to prepare high molecular weight block copolymers.
First Block:
MMA/CDB/CuBr/Cu0/TPMA = 2,000/1/0.01/5/0.03 in anisole 66 (v/v) %, 32 h @ 80 oC
Conversion = 61.1%; Mnth =123,000 g/mol; Mn = 112,000 g/mol; Mw/Mn = 1.08
Second Block:
BMA/PMMA/CuBr/Cu0/TPMA = 30,300/1/0.01/5/0.03 in anisole 50 (v/v) %, 92 h @ 80 oC
The molecular weight of the second block reached 1,400,000 after 92 hours and Mw/Mn = 1.2.
REFERENCES
(1) Davis, K. A.; Matyjaszewski, K. Advances in Polymer Science 2002, 159, 2-166.
(2) Li, M.; Jahed, N. M.; Min, K.; Matyjaszewski, K. Macromolecules 2004, 37, 2434-2441.
(3) Matyjaszewski, K. Polymer International 2003, 52, 1559-1565.
(4) Xia, J.; Paik, H.-j.; Matyjaszewski, K. Macromolecules 1999, 32, 8310-8314.
(5) Coca, S.; Matyjaszewski, K. J. Polym. Sci., Part A: Polym. Chem. 1997, 35, 3595-3601.
(6) Jakubowski, W.; Tsarevsky, N. V.; Higashihara, T.; Faust, R.; Matyjaszewski, K. Macromolecules 2008, 41, 2318-2323.
(7) Acar, M. H.; Matyjaszewski, K. Macromol. Chem. Phys. 1999, 200, 1094-1100.
(8) Coca, S.; Paik, H.-j.; Matyjaszewski, K. Macromolecules 1997, 30, 6513-6516.
(9) Gaynor, S. G.; Matyjaszewski, K. Macromolecules 1997, 30, 4241-4243.
(10) Inoue, Y.; Matyjaszewski, K. Journal of Polymer Science, Part A: Polymer Chemistry 2004, 42, 496-504.
(11) Hong, S. C.; Lutz, J.-F.; Inoue, Y.; Strissel, C.; Nuyken, O.; Matyjaszewski, K. Macromolecules 2003, 36, 1075-1082.
(12) Matyjaszewski, K.; Shipp, D. A.; McMurtry, G. P.; Gaynor, S. G.; Pakula, T. J. Polym. Sci., Part A: Polym. Chem. 2000, 38, 2023-2031.
(13) Kowalewski, T.; Tsarevsky, N. V.; Matyjaszewski, K. Journal of the American Chemical Society 2002, 124, 10632-10633.
(14) Kowalewski, T.; Tang, C.; Kruk, M.; Dufour, B.; Matyjaszewski, K. ACS Symposium Series 2006, 944, 295-310.
(15) Dufour, B.; Tang, C.; Koynov, K.; Zhang, Y.; Pakula, T.; Matyjaszewski, K. Macromolecules 2008, 41, 2451-2458.
(16) Matyjaszewski, K.; Ziegler, M. J.; Arehart, S. V.; Greszta, D.; Pakula, T. J. Phys. Org. Chem. 2000, 13, 775-786.
(17) Angot, S.; Murthy, K. S.; Taton, D.; Gnanou, Y. Macromolecules 1998, 31, 7218-7225.
(18) Kolb, H. C.; Finn, M. G.; Sharpless, K. B. Angewandte Chemie, International Edition 2001, 40, 2004-2021.
(19) Golas, P. L.; Tsarevsky, N. V.; Sumerlin, B. S.; Matyjaszewski, K. Macromolecules 2006, 39, 6451-6457.
(20) Golas, P. L.; Tsarevsky, N. V.; Sumerlin, B. S.; Walker, L. M.; Matyjaszewski, K. Australian Journal of Chemistry 2007, 60, 400-404.
(21) Zarafshani, Z.; Akdemir, O.; Lutz, J.-F. Macromol. Rapid Commun. 2008, 29, 1161-1166.
(22) McCormick, C. L.; Sumerlin, B. S.; Lokitz, B. S.; Stempka, J. E. Soft Matter 2008, 4, 1760-1773.
(23) Nicolay, R.; Kwak, Y.; Matyjaszewski, K. Chemical Communications (Cambridge, United Kingdom) 2008, 5336-5338.