Electrochemical control over an ATRP
eATRPPotentiostatic versus Galvanostatic Conditions
Simplified eATRP (seATRP)
eRAFT
eATRP Emulsion Polymerization
eATRP
When one considers the status of ATRP at the end of 2010, after exemplification of ARGET and ICAR ATRP (1) and an ab initio emulsion ATRP process, (2) one could ask what targets remain in order to make ATRP more industrially compatible? The answers include, as always: reduce costs, elimination/reduction of reagents, define conditions that allow simplified reaction conditions e.g. low levels of O2 permissible and, if possible provide additional degrees of polymerization control.So what was next?
It was a new environmentally friendly procedure for controlling the ratio of activator to deactivator in an ATRP, specifically by electrochemical procedures. Electrochemical methods offer multiple readily adjustable parameters, e.g. applied current, potential, and total passed charge, that can be applied to manipulate polymerization rates by selective targeting of specific relative concentrations of redox-active catalytic species which will provide industrially scalable procedures that allow for the creation of more complex specialized materials.
Cyclic voltammetric (CV) studies of copper complexes suitable for catalyzing atom transfer radical polymerization have been used since ~2000 to measure the relative activity of copper based catalyst complexes in an ATRP. (3, 4) It was found that the E1/2 value for the redox couple CuI/II/L strongly depends on the nature of the ligand and the selected transferable halogen, Figure 1.
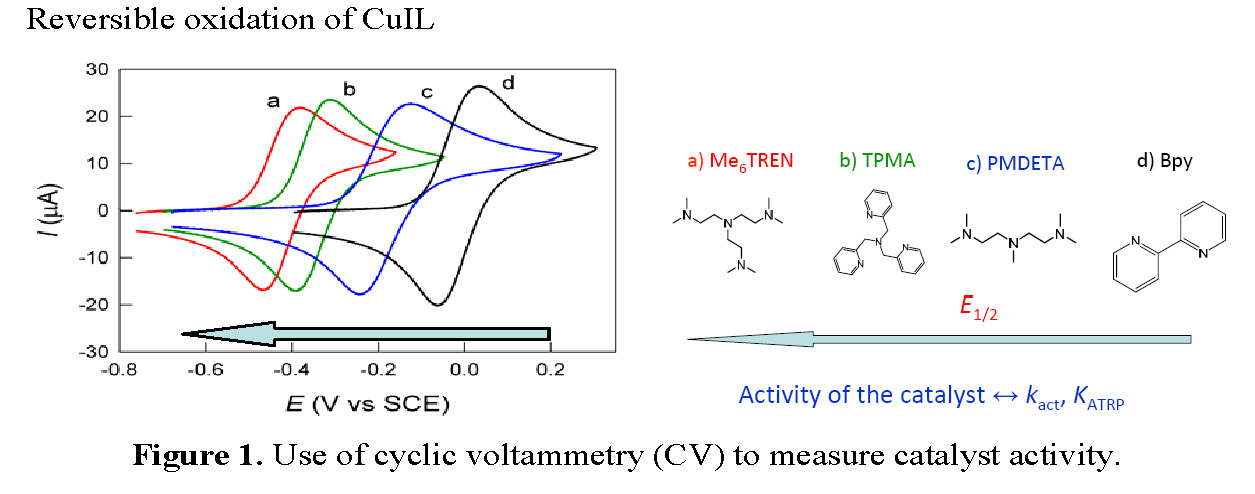
An initial report exploring the possibility of using an electrochemically-triggered initiation of controlled polymerization sequence during a CV cycle involving activation of alkyl- and benzyl halide initiators by an electrogenerated FeIISalen complex was reported in 2009. (8) The reactions between the formed FeIISalen and several organic halides were investigated within the time scale of cyclic voltammetry measurements. A fast reaction was observed between electrogenerated FeIISalen catalyst complex and ATRP initiator molecule in the presence of styrene at 110 °C. Polymerization was initiated and the polydispersity of the resulting low molecular weight polystyrene appeared to depend on the initial FeIIISalen/FeIISalen ratio. However this work was apparently not continued or expanded.
The proposed mechanism of ATRP mediated through electrochemical (re)generation of activators is shown below in Figure 2.

Figure 2. Schematic of proposed mechanism for electrochemical control over an ATRP
The schematic demonstrates the dynamic modulation of polymerization rates through electrochemical means, essentially a specific targeted fraction of the air stable CuIIBr2/Me6TREN catalyst complexes can be electrochemically reduced at the working electrode under cathodic currents to form the CuIBr/Me6TREN activator that can invoke or trigger a controlled polymerization in the presence of the remaining CuIIBr2/Me6TREN catalyst complexes. In absence of mass transport limitations, the rate of reduction is dictated by the applied potential (Eapp) allowing one to finely tune the polymerization rate by balancing the ratio of [CuI]/[CuII] and thereby fine tuning of the polymerization rate. Further to this point, electrochemical methods allow the lower oxidation state catalyst, exemplified in Scheme 2 by CuIBr/Me6TREN, to be reverted back to its original higher oxidation state, simply by shifting Eapp to more positive values and in doing so providing a means to deactivate an ongoing polymerization and thereby control any temperature exotherms. This dormant time in the polymerization may also be used for modification of the polymer or injection of additional reagents/monomers. This procedure for an electrochemically mediated ATRP has been given the abbreviation eATRP.
The initial paper that actually studied procedures for electrochemical control over an ATRP (9) demonstrated that the dynamic modulation of polymerization rates through electrochemistry provided a means to allow "on demand" polymerization initiation, cessation of polymerization, and rejuvenation of a controlled/living radical polymerization process. Applying electricity to the system provided more precise control over the reaction. Electrochemical methods provide almost instantaneous control over an ATRP by offering additional adjustable control parameters, e.g. current, potential, and total passed charge, to manipulate polymerization activation/deactivation and polymerization rates by selective targeting of a desired ratio of redox-active catalytic species. A computer-controlled battery allows one to manipulate the ATRP process in real-time by changing the current or voltage. Moreover, all the advantages of low catalyst concentration ATRP procedures, such as ARGET ATRP, are maintained by achieving ppm concentrations of catalyst (1, 10) and tolerance to ambient O2, (11) while simultaneously offering an environmentally friendly alternative by elimination of chemical reducing agents in addition to providing a convenient catalyst removal/recycle process through electrodeposition. (7)
A schematic and an image of the equipment used for this initial eATRP study are shown in Figure 3.
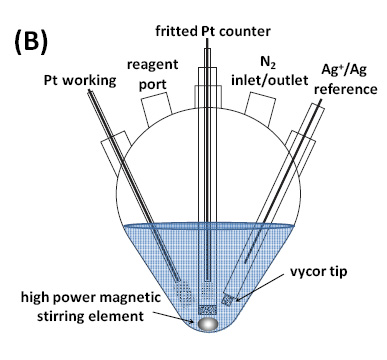

Figure 3. Five necked flask with three electrodes used for e-ATRP: the Pt working electrode, the counter electrode, and the reference electrode which were a 3 mm Pt disk (Gamry Instruments), a Pt mesh, and an Ag|Ag+, which contained a 0.1 M AgNO3 and TBAPF6 filling solution in MeCN separated from the working solutions by a porous Vycor tip, respectively.
In an eATRP, a desired amount of the catalyst complex (X-CuII/L) can be electrochemically reduced to activators (CuI/L) to start a controlled radical polymerization. (9) Typically, the reaction mixture initially contained solvents, monomers, initiators, supporting electrolyte and X-CuII/L. Due to absent of CuI/L activators, polymerization has not occurred. The polymerization begins only when a sufficient cathodic electrical current was applied to the working electrode (WE) for reduction of a fraction of the added X-CuII/L to CuI/L at the WE surface. The reduced activators then spreads out into the working solution by vigorous stirring, and reacts with initiators (e.g., alkyl halide, Pn-X) to form radicals (Pn•) and is oxidized back to deactivators (X-CuII/L). The radical species propagated to form polymeric chains by reacting with monomers (M), or were deactivated back to the dormant species (Pn-X), Figure 2. Continuous (re)generation of activators (CuI/L) and modulation of the rate of polymerization (Rp) can be achieved by an appropriately selected Eapp.
As noted above, applying the appropriate charge is a requirement for a well-controlled electrochemical ATRP reaction. In the initial example reduction of the CuIIBr2/Me6TREN complex was accomplished through application of a cathodic current. Prior to the electrolysis experiments, cyclic voltammograms (CVs) of methyl acrylate (MA) and acetonitrile (MeCN) over a potential range of 1.5 volts, from 0.0 to -1.5 V versus Ag|Ag+ reference electrode were acquired to ensure the absence of any redox process that might interfere with the catalyst reduction. After confirming the electrochemical stability of the reaction medium an additional CV in the presence of added CuIIBr2/Me6TREN was acquired to identify the potential window appropriate for accurate manipulation of the oxidation states of the redox-active catalyst in electrolysis experiments. A reversible peak couple attributed to the single electron transfer reduction of the X-CuII/L complex was observed, as shown in Figure 4. The value of the half wave potential of the copper catalyst (CuI/II/L) was calculated to be E1/2 = -0.69 V versus Ag/AgI/I-.
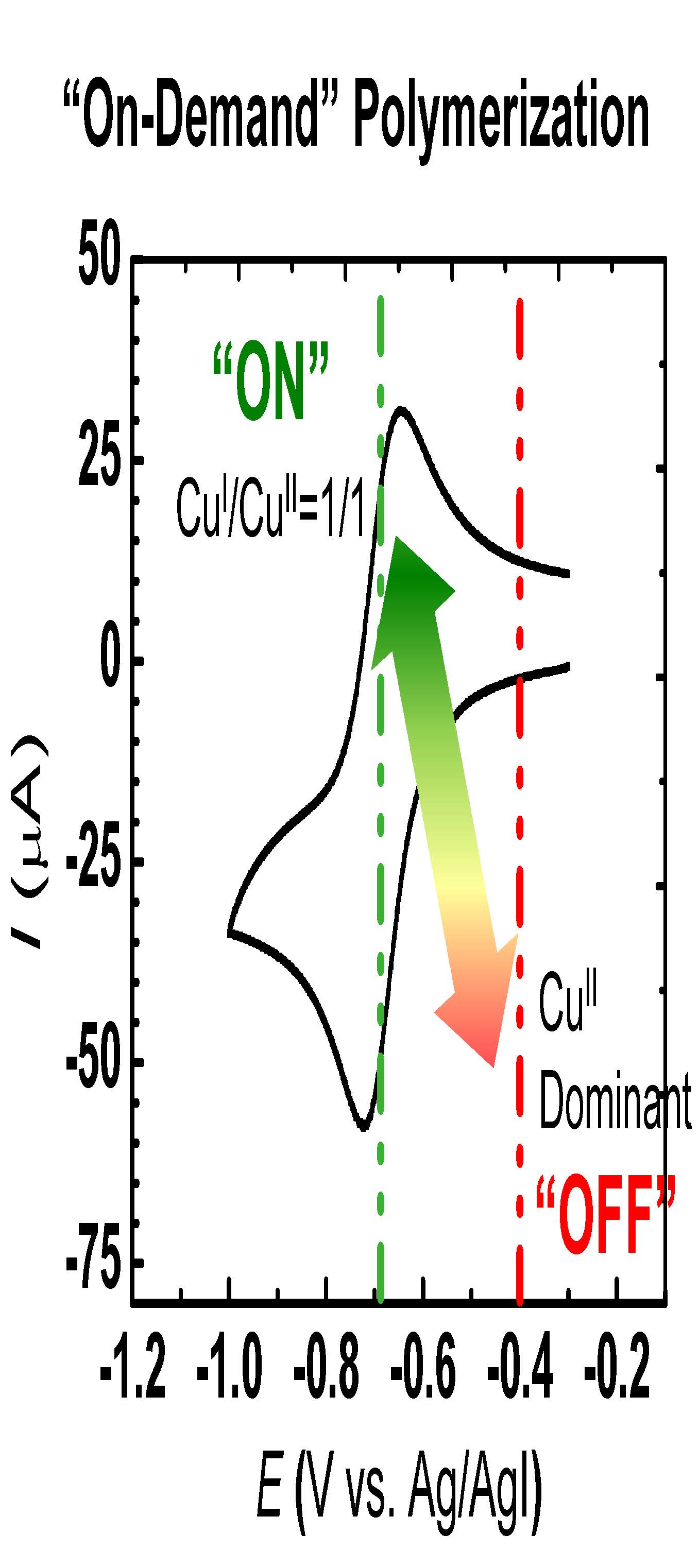
Figure 4: Cyclic voltammetry of 1.0 mM CuIIBr2/Me6TREN recorded at v = 0.5 V/s-1 in 50 % (v/v) MA in MeCN with 0.1 M supporting electrolyte.
The application of a -0.66 V potential to the reaction medium resulted in the rapid consumption of monomer, reaching nearly 80 % conversion within four hours, Figure 5A blue points. Linear first order kinetic behavior was observed during the polymerization, Figure 5B, indicating a constant concentration of propagating species, and an excellent correlation persisted between theoretical and experimental molecular weight values as the polymerization progressed. Molecular weight values increased linearly with conversion and molecular weight distributions decreased gradually with increasing monomer conversion reaching a minimum value of approximately Mw/Mn = 1.06. Since the rate of an ATRP is defined by ratio of [CuIBr/Me6TREN] and [CuIIBr2/Me6TREN], and therefore depends on the extent to which CuIIBr2/Me6TREN is reduced and consequently the Eapp.
The choice of the Eapp is of primary importance in a polymerization conducted under potentiostatic conditions. Specifically, two conditions can be used to control the reaction; the first is electrolysis under diffusive control, where Eapp is more negative than Epc and the overall rate of the reaction is controlled by the rate of the diffusion of the reactants to the electrode surface rather than the rate of the reaction itself. The second is electrolysis controlled by the rate of electron transfer, in this case Eapp is more positive than Epc and the overall rate of the reaction is controlled by the rate of the reaction itself rather than the rate of the mass transport of the reactants to the electrode surface. In addition to these considerations, the selected Eapp should avoid other undesired electron transfer (ET) reactions. In ATRP systems, one of these undesired ET reactions is the further reduction of CuI/L to Cu0. This is expected to occur at potentials more negative than that of the CuI/II/L redox coupling potential when disproportionation is not thermodynamically favoured. (12) It should be selected only when one desires to remove the copper from the reaction medium.
Two additional and separate polymerizations were conducted with a 30 and 60 mV increase of the Eapp to -0.69 and -0.72 V, respectively. An enhanced polymerization rate was observed at more negative potentials whereas more positive potentials provided a slower rate of polymerization, Figure 5A.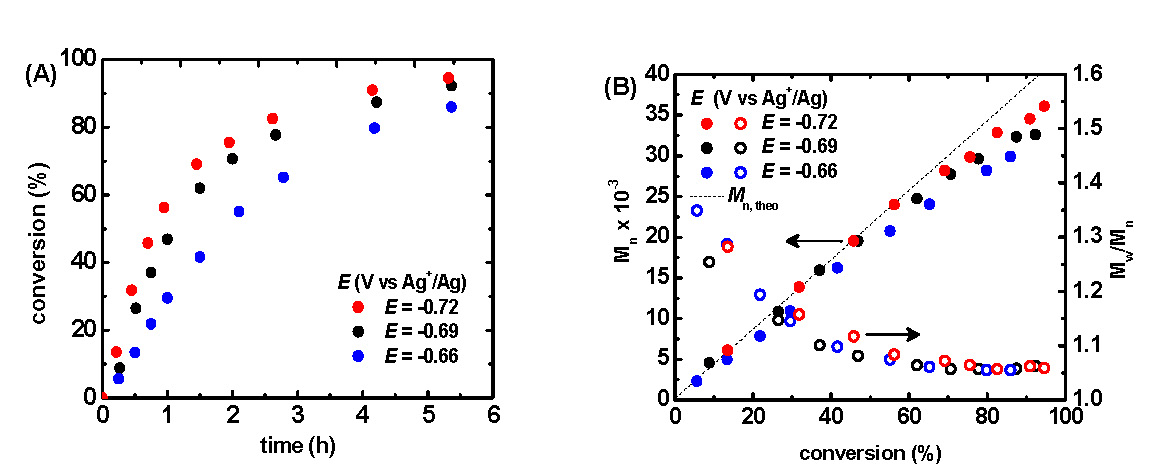
Figure 5:(A) Monomer conversion with respect to time, and (B) number average molecular weight (Mn) and Mw/Mn with respect to conversion as a function of applied potential. Polymerizations conducted in 50% (v/v) MA in MeCN at 25° C with a total reaction volume of approximately 12 mL. [MA]0 = 5.55 M; [MA]0:[EBP]0:[Me6TREN]0:[CuIIBr2]0 = 500:1:0.025:0.025.
A high cathodic decay has been observed in the early stages of a polymerization conducted under potentiostatic conditions (constant potential), representing the reaction of the reduced CuI/L complex with an initiator followed by an EC' (electrolysis-catalysis) mechanism. More negative η produces larger initial currents, indicating a faster rate of reduction. Subsequently, a steady-state condition, equilibria of [Pn-X], [Pn•], [CuI/L], and [X-CuII/L] is established, hence constant current is observed. (13) This paper also examined the effect of stirring and the effect of distance from the reducing electrode as the polymerization kinetics depend on the ratio of [CuII]/[CuI]. The ratio can be adjusted by varying the applied potentials with fast stirring or the distance (d) between the working electrode and the initiator modified surface (without stirring). In the first case, the reduction of the catalysts only occurs at the WE surfaces and fast stirring efficiently spreads out the reduced activators and prevents passivation of the growing polymer chains at the WE surfaces if stirring is diminished or stopped. The influence of the distance will be addressed below when discussing the preparation of polymeric surfaces by surface-initiated eATRP.
As with all other ATRP procedures, catalyst complexes with higher reducing power provide faster polymerizations, due to larger value for KATRP. eATRP is also influences by the supporting electrolyte and a decrease in rate was observed when the concentration of electrolyte was decreased. This decrease can be explained as a consequence of an increase in the resistance of the contacting solution. When all processes involved in the reaction are fast then electron transfer is the limiting factor in the reaction. (12)
Typically eATRP reactions were carried out in DMF and MeCN as solvents with polymerizations fasted in DMF due to the influence of polarity on KATRP. For instance KATRP is ~103 times higher in aqueous media than in MeCN. As noted elsewhere on the site eATRP overcomes many of the drawbacks typically associated with conducting an ATRP protic media (14) and recently water soluble methacrylates and acrylamides have been successfully polymerized by eATRP providing polymers with narrow molecular weight distribution and targeted degrees of polymerization. (14-18) The X-CuII/L bond can easily dissociate in an aqueous ATRP, therefore either high concentrations of catalysts or addition of salts (halide anions, X-) were required. (14, 19) The addition of excess X- promotes the (re)formation of X-CuII/L and a noticeable improvement of MW distribution indeed targeting conditions that result in formation of a constant and high [CuII]/[CuI] ratio is optimal. Reaction temperature also exerts an influence in an aqueous ATRP as concurrent nucleophilic substitution (solvolysis) of alkyl halides in water can be suppressed at lower temperatures. Thus, aqueous systems that are conducted at relatively low temperature and high concentration of Cu catalysts seem to be most effective. (15, 20, 21)
Potentiostatic versus galvanostatic conditions
Electrolysis under galvanostatic conditions is attractive from both an industrial and experimental standpoint because it provides a constant current instead of a constant potential and eliminates the need for the reference electrode (RE). Galvanostatic conditions have been successfully utilized for the eATRP synthesis of homopolymers and copolymers. (13)
eATRP reactions were conducted under galvanostatic or potentiostatic conditions and Figure 6 shows a comparison of the applied conditions and results. In this case, the selected galvanostatic current values resembled those of the potentiostatic polymerization (Q = A × s), whereby an initial high current value, red section Figure 6(a), and consecutive low current value, blue section Figure 6(a), were employed. This two-stage current program was chosen to rapidly convert the vast majority of X-CuII/L to CuI/L, initiating eATRP, and next lower current step providing compensation for any (re)generated X-CuII/L incurred from termination.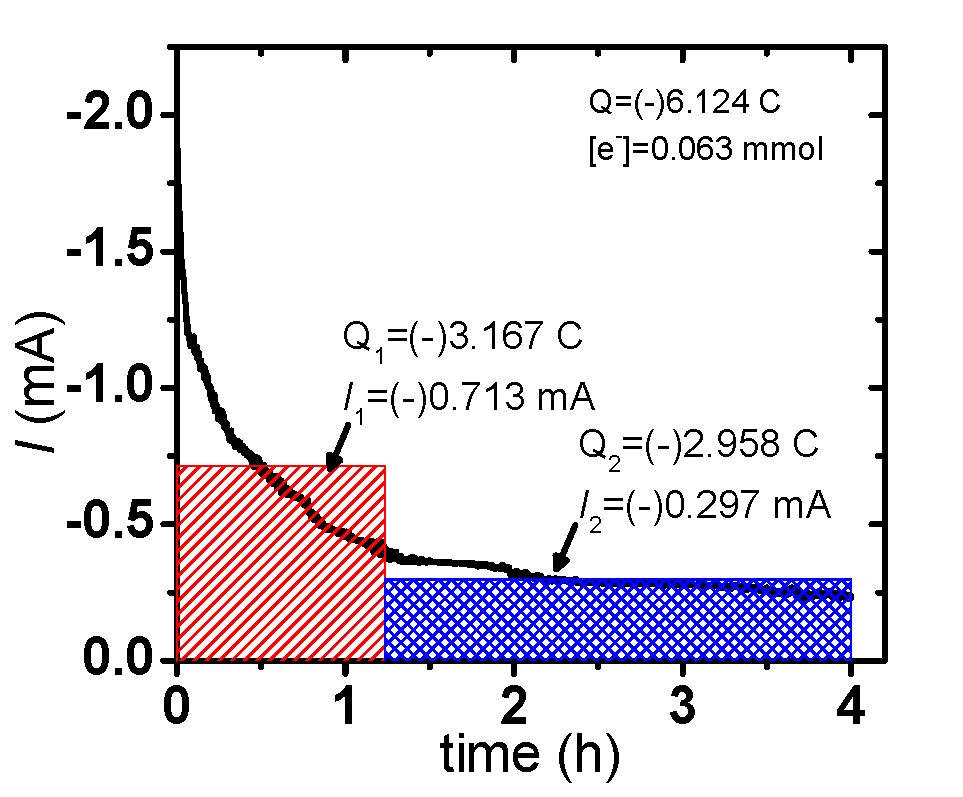
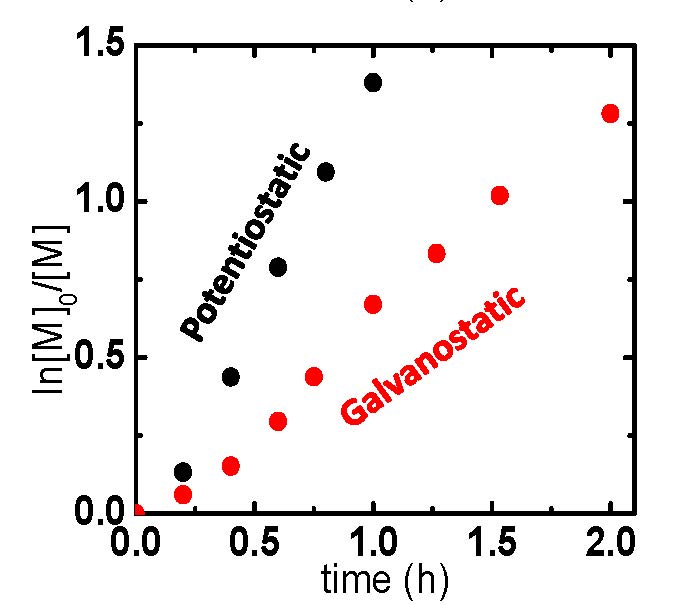
Figure 6. eATRP of n-butyl acrylate (BA): (a) Current profiles of (black) a potentiostatic (black) and (red/blue) galvanostatic polymerization. (b) First-order kinetic plot of a potentiostatic and galvanostatic eATRP.
As the polymerization took place the potential remained in close proximity to the potential of Cu/L couple, however, near the end of each current stage it began to shift to more negative potentials. This indicates that in the vicinity of the electrode, nearly all X-CuII/L was converted to CuI/L, and therefore the potentiostat began to apply a stronger reducing potential to maintain these current values. The polymerization under galvanostatic conditions was slower than the polymerization under the potentiostatic setup, Figure 6(b), because of a lower applied current, i.e. a smaller amount of charge (Q) passed at the same time period. With regards to polymerization control, the polymerization displayed a linear increase in MW with conversion and low Mw/Mn values during the course of the polymerization
Simplified eATRP (seATRP)
The complexity of an electrochemical reaction setup can be overcome by using sacrificial anode (counter electrode). The simplified eATRP (seATRP) does not require any separation of CE from the reaction medium and can be directly inserted into the reaction mixture, Figure 7.
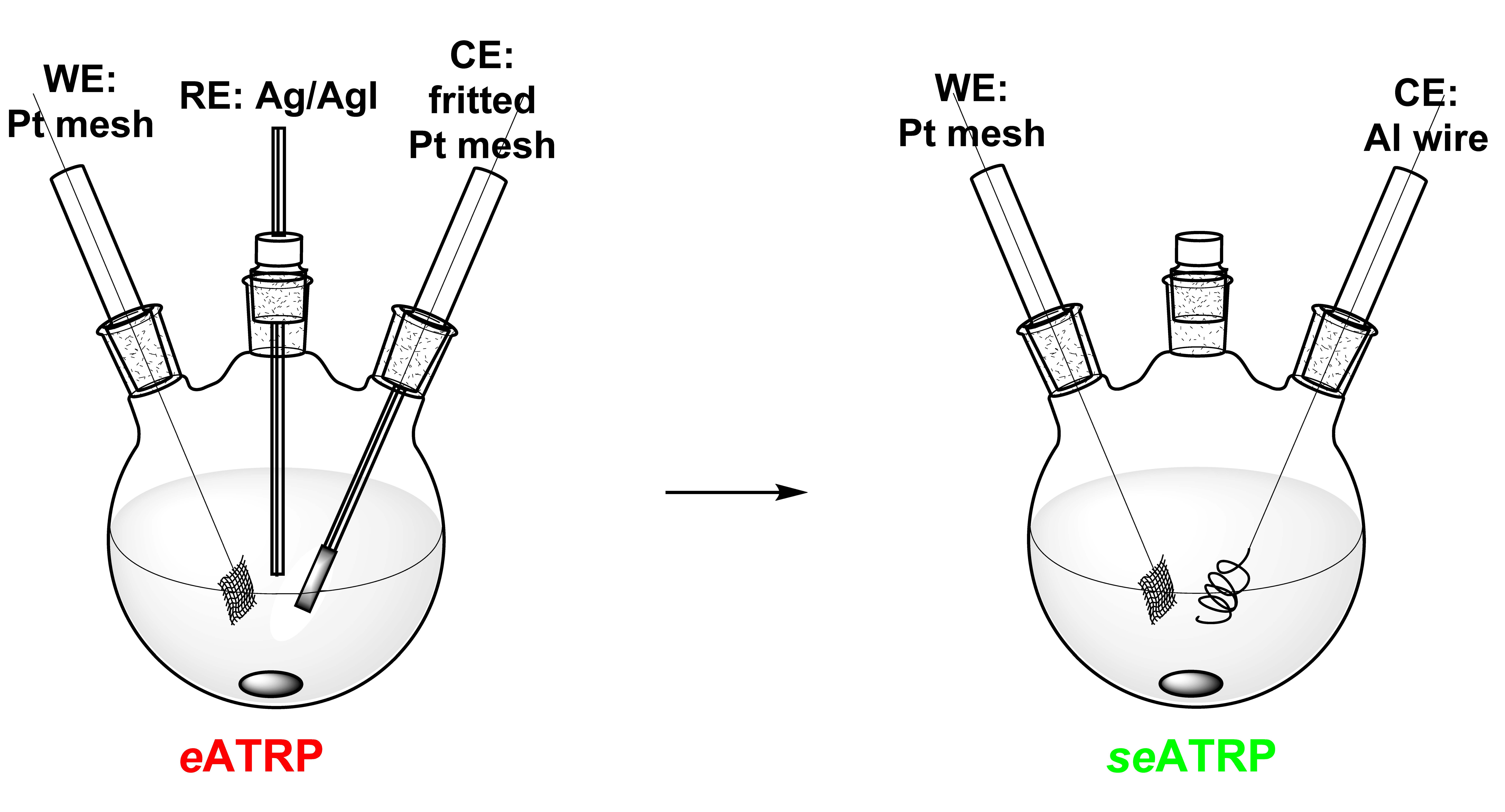
Figure 7. Comparison of setup required for an eATRP and a seATRP
There are two potential advantages of using seATRP. The first one is the use of an undivided cell, which allows for a simpler, lower cost process, and most of all, the minimization of the ohmic drop, bringing a beneficial saving in energy costs. The second advantage is the possibility of a galvanostatic process using two electrode system and a power supply, which is simpler than a potentiostatic process. A series of seATRP polymerizations were carried out under different Eapp, with a Pt mesh WE, an aluminum (Al) wire CE, and Ag/AgI/I- RE. (22) In the ideal case of seATRP, the sacrificial counter electrode as well as the products of its oxidation should not react with the CuI/L catalysts. In this case, aluminum (Al) does not reduce X-CuII/L to CuI/L even though it usually shows a negative standard potential in different media. This is likely because the surface of the Al is passivated by forming stable oxidized layers to prevent reduction of X-CuII/L to CuI/L. Therefore the Al seemed to fit the requirements of a sacrificial electrode. The polymerization results showed similar MW evolution, maintaining a narrow MWD throughout the reactions which are similar to results observed for n-butyl acrylate (BA) polymerization under conventional eATRP. In addition, galvanostatic conditions with a two electrode system were developed for synthesizing PBA. The direct immersion of an Al wire anode in the reaction medium can avoid additional preparation setup steps, and the polymerization results indicated good control of reaction kinetics, providing polymers with molecular weight evolution close to theoretical values and generating polymers with narrow molecular-weight distribution. The rate of the polymerizations Rp was controlled by applying different potentials Eapp, with faster Rp observed using more negative Eapp. Synthesis of high MW polymers and chain extension reactions indicated good conservation of chain-end functionalities. The seATRP procedure can be further simplified by using only two electrodes and applying a constant current under galvanostatic conditions. The use of a multi-step current procedure showed identical results to polymerizations carried out under potentiostatic conditions, namely linear first-order kinetics and a uniform growth of polymers.
Electrochemically Mediated Reversible Addition-Fragmentation Chain-Transfer (eRAFT) Polymerization
Another important RDRP procedure is RAFT polymerization which is based on degenerative transfer, and is mediated by chain transfer agents (CTAs) such as dithioesters, dithiocarbamates, trithiocarbonates, or xanthates. RAFT polymerization is compatible with many vinyl monomers and solvents, including both homogeneous and
heterogeneous conditions. However in a RAFT polymerization a continuous supply of radicals (i.e., “initiation”) is essential. Initiation can be accomplished by thermal decomposition of radical sources such as azobis(isobutyronitrile) (AIBN) between 50 and 70 °C. However, such temperatures may be not suitable for some functional monomers or for the preparation of biological or thermoresponsive systems.
Direct electroreduction of chain transfer agents was unsuccessful, since it resulted in the formation of carbanions by a two-electron-transfer process. An electrochemically mediated RAFT (eRAFT) polymerization of (meth)acrylates was successfully carried out via electroreduction of either benzoyl peroxide (BPO) or 4-bromobenzenediazonium tetrafluoroborate (BrPhN2+), which formed aryl radicals, acting as initiators for RAFT polymerization. (23) Electroreduction of BPO initiated a conventional free radical polymerization and was used for a successful eRAFT at potentials Eapp ≈ Epc,BPO. Since the reduction of BPO partially overlapped with reduction of the CTAs this limited the use of the peroxide for eRAFT polymerization. Well-defined polymers were prepared by applying either fixed potential or fixed current conditions. eRAFT with BrPhN2+ can be carried out under galvanostatic conditions at a single current value, utilizing a simple current generator at ambient temperature. The amount of new chains generated by reduction of BrPhN2+ was lowered by decreasing Iapp or by decreasing the amount of diazonium salt relative to the CTA.
Lower Iapps produced better controlled polymers, but at slower rates, and conversions >80% were achieved. The chain-end functionality was well-maintained, as confirmed by synthesis of well-defined homopolymers and block copolymers with a broad range of targeted degrees of polymerization.
eATRP in Emulsion Polymerization
eATRP in Miniemulsion with a Dual Catalytic System
eATRP in Miniemulsion with a Single Catalytic Complex
Dispersed media, such as miniemulsions, are useful eco-friendly systems for the preparation of functional copolymers as the process is less toxic, and less expensive than most organic solvents. In particular, oil-in-water miniemulsions have many applications, enabled by the submicrometer size of the dispersed oil droplets, by the large array of possible hydrophobic reactants, and by the excellent mass and heat transport properties of the aqueous suspension medium. Combining eco-friendly (mini)emulsions with electrosynthetic methods offers an additional advantage, since the use of electrons as reagents does not involve the formation of side products observed when using reducing or oxidizing agents for catalyst activation.
eATRP in Miniemulsion with a Dual Catalytic System
An electrochemical approach was used to control an ATRP of n-butyl acrylate (BA) in miniemulsion. (24) Initially it was believed that electropolymerization required a dual catalytic system, composed of an aqueous phase catalyst and an organic phase catalyst thereby allowing shuttling the electrochemical stimulus from the working electrode (WE) to the continuous aqueous phase and to the dispersed monomer droplets where they can interact with the hydrophobic catalyst complex in the dispersed droplets and activate the polymerization. Hydrophilic Cu complexes with N,N-bis(2-pyridylmethyl)-2-hydroxyethylamine (BPMEA), 2,2′-bipyridine (bpy), and tris(2-pyridylmethyl)amine (TPMA) ligands were tested as the aqueous suspension phase catalysts while hydrophobic complexes with either bis(2-pyridylmethyl)-octadecylamine (BPMODA) or bis[2-(4-methoxy-3,5-dimethyl)-pyridylmethyl]octadecylamine (BPMODA*) ligands were evaluated as the organic phase catalysts.
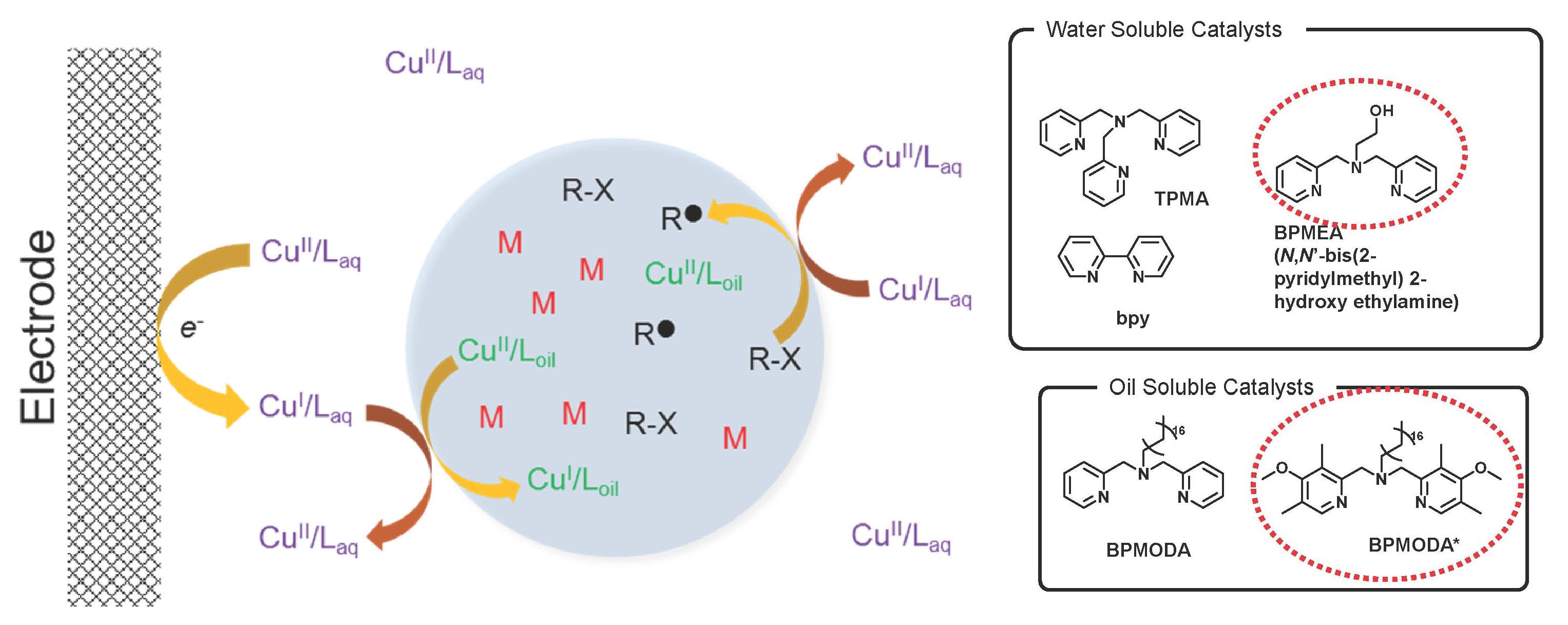
Highest rates and best control of BA electropolymerization were obtained with the water-soluble Cu/BPMEA used in combination with the oil-soluble Cu/BPMODA*. The polymerization rate could be further enhanced by changing the potential applied at the WE. In contrast to the results from traditional ATRP systems, reactivity of the dual catalytic system did not depend on the redox potential of the catalysts but instead depended on the hydrophobicity and partition coefficient of the aqueous phase catalyst. The proper balance between redox potentials and hydrophilic/hydrophobic properties of the dual catalytic system provided efficient electrocatalysis and yielded well-defined polymers by eATRP in miniemulsion.
Polymerizations showed almost linear first-order kinetics generating polymers with Mn matching the theoretical values, and low Ð. In each case, GPC traces showed a clear peak shift from low to high molar mass. Polymerization rate was influenced by the concentration of initiator and the lowest target DP showed the highest apparent rate of polymerization (kpapp). Eapp was selected from the reversible CV recorded in the polymerization mixture. For each of the three selected Eapps, Mn increased linearly with monomer conversion and matched well the theoretical values (Mn,th). A narrow molecular-weight distribution was obtained, indicating uniform growth of polymer chains. As expected, more reducing conditions resulted in an increased reaction rate: kpapp gradually increased with decreasing Eapp (kpapp = 0.035, 0.036, and 0.041 h-1 for E1/2, Epc, and Epc – 80 mV, respectively. The most reducing conditions led to the highest monomer conversion after 24 h (71% at Eapp = Epc – 80 mV, approximately the mass transport limit for CuBr2/Laq reduction). In contrast to examples from traditional ATRP systems, the reactivity of the dual catalytic system did not depend on the redox potential of the catalysts, but instead depended on the hydrophobicity and partition coefficient of the aqueous phase catalyst.
This then led to the question of whether a single catalyst could be employed to conduct an eATRP miniemulsion polymerization?
eATRP in Miniemulsion with a Single Catalytic Complex
In order to make such a concept work it would be necessary to select an aqueous phase catalyst which had direct contact with the WE and additionally possessed the ability to migrate to monomer droplets and interact with the initiator/growing chain end within the dispersed droplets. This would require good selection of the catalyst complex. A series of runs were conducted and the best result was observed for a reaction conducted with TPMA as ligand.
The next step was investigation of migration of the copper catalyst and the influence of surfactants on the migration procedure and surprisingly it was observed that SDS formed an ionic complex with the catalyst that assisted in transfer of the catalyst complex into the oil phase. (25) It was discovered that if the cationic copper complex is paired with an anionic surfactant. The catalyst/surfactant system then generates ion pairs that transport the catalyst into the hydrophobic monomer droplets, thus allowing a controlled polymerization.
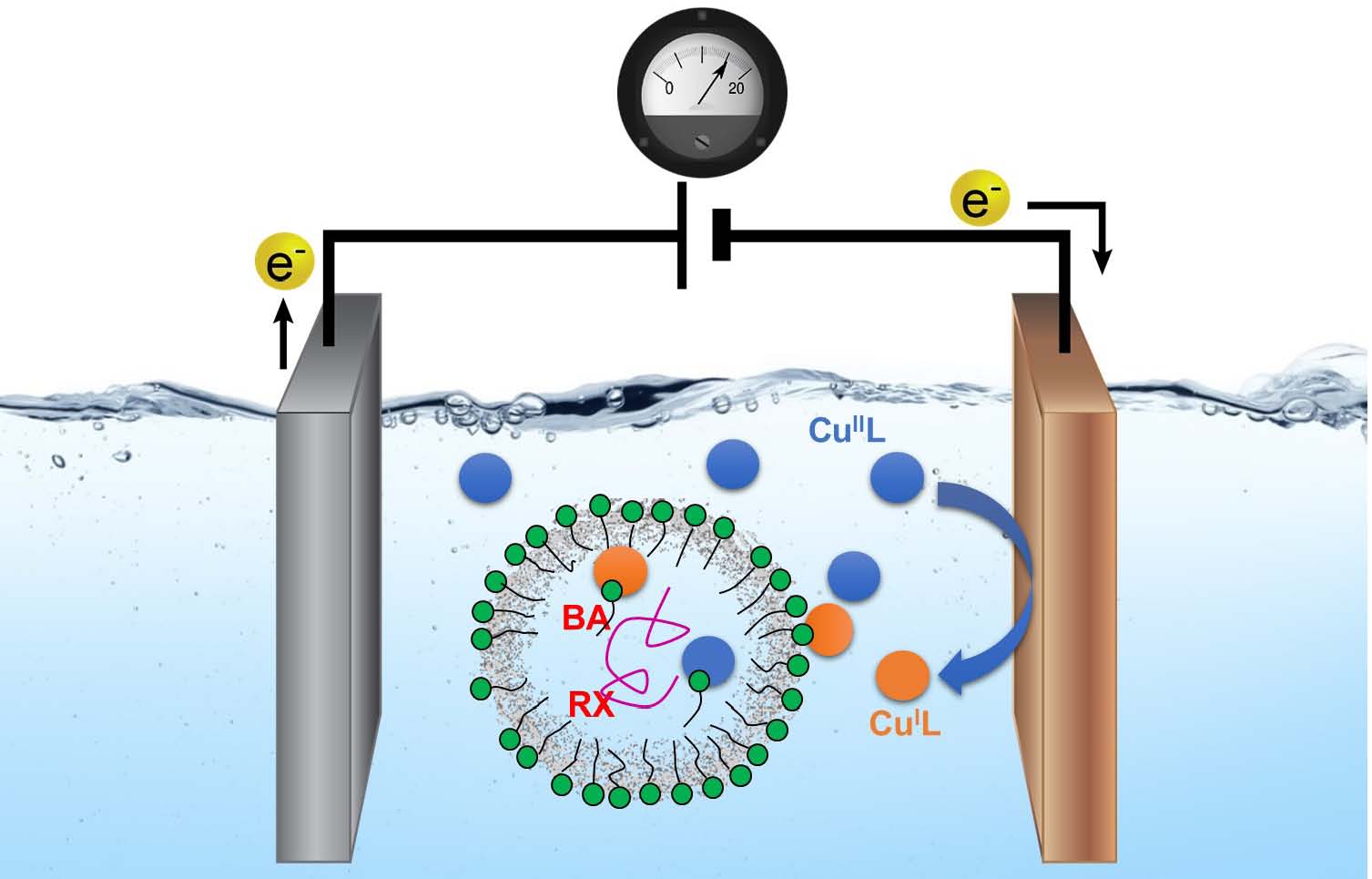
Historically nonionic surfactants were typically employed in miniemulsion ATRP and until recently, anionic surfactants such as sodium dodecyl sulfate (SDS) were considered a poison for ATRP catalysts (26) since the surfactant (DS− anion) can coordinate to CuIIL+, displacing Br− from the deactivator Br−CuIIL+. However it was determined (27) that if excess NaBr (0.1 M) was added to the reaction medium this undesired reaction was avoided. Under such conditions, Br−CuIIL+ was stable and was therefore considered the main CuII species present in the aqueous phase. It was reported that a successful miniemulsion ATRP could be conducted using an anionic surfactant by minimization of deactivator loss by addition of a halide salt. We determined that the interaction between SDS and the CuII/L complex (L= amine ligand) could be exploited to increase the concentration of the catalyst in the polymerization loci. (25)
Unexpectedly, it was observed that the superactive catalyst Br−CuIITPMA*2+ provided a slower polymerization rate and poorer control than the complex formed with TPMA, indicating that catalyst distribution and interfacial dynamics may play a more important role than the thermodynamics of RX activation.
The efficiency of the Br−CuIITPMA+/SDS complex catalyst was tested over a range of catalyst concentrations, 1200−300 ppm of Cu. Controlled eATRP was obtained in each case, with a linear increase of molecular weight (MW) with conversion. However, polymerization rate tended to slow down at high conversion in the presence of a lower concentration of catalyst. Higher catalyst loadings provided higher rates of polymerization and higher conversions with narrower Đ.
The interaction between the catalyst complex and SDS was examined by CV. CV was first applied to a model heterogeneous system, composed of catalyst and surfactant, but without the presence of monomer. In this medium, micelles with diameter ca. 2 nm were formed, as compared to larger miniemulsion droplets with diameter ca. 100 nm. The CV of Br−CuIITPMA+ transition metal complex in water drastically changed after addition of SDS. The lower current indicated that the diffusion coefficient (D) of Br−CuIITPMA+ diminished, because the catalyst was bound to the surface of much larger micelles, which diffused slowly throughout the solution.
A similar behavior was observed for other CuII complexes. Another feature of the CV in the presence of SDS is a −0.03 V shift of the half-wave potential (E1/2). This indicated that the CuII deactivator complex had ca. 3 times more affinity for the micellar environment than the CuI activator complex. This is an important aspect of the disclosed procedure as the higher concentration of CuII can enhance deactivation and control an ATRP conducted within the distributed micelles. Indeed, the poor polymerization control obtained with Br−CuIIMe6TREN+ could be due to its weaker interaction with SDS, which favored activation over deactivation.
The fraction of Br−CuIITPMA+ bound to SDS stabilized micelles (fbound) was determined by comparing the diffusion coefficient (D) of Br−CuIITPMA+ in pure water to its D in the presence of micelles. The value for fbound of Br−CuIITPMA+ was similar in micellar and in miniemulsion environments: ≥79% of the catalyst was bound to the interface, confirming the strong interaction between Br−CuIITPMA+ and SDS. This analysis indicated that a large fraction of Br−CuIITPMA+ was present at the droplets’ surface, from where it could activate and deactivate the growing chains by an interfacial catalysis procedure. However, such heterogeneous catalysts are typically inefficient deactivators which would lead to a broad Đ. Therefore, we investigated if, in the presence of SDS, some of the catalyst could be transported inside the monomer droplets, acting as a traditional homogeneous catalyst and supplementing the interfacial catalysis. Vis-NIR in BA phase indicated that ~1% of Br-CuII/L was present within the droplets.
It was determined that a miniemulsion eATRP can be carried out with an anionic surfactant and a single, strongly hydrophilic catalyst. The obtained PBA was well-defined, with MWs matching theoretical values and low Đ. Furthermore, chain extension experiments confirmed that chain-end functionality was very well retained which was confirmed by two different chain extension experiments using a PBA78-Br macroinitiator prepared by simplified electrochemically mediated ATRP (seATRP) in miniemulsion. The following figures show (A) MW and Đ vs monomer conversion. (B, C) GPC traces. The schematics shows the results of the experiments with in situ chain extension in red and blue = chain extension after purification of the PBA-Br macroinitiator. Reaction conditions were [tBA]/[PBA78-Br]/[CuII] = 80/1/0.06, 20% v/v tBA.
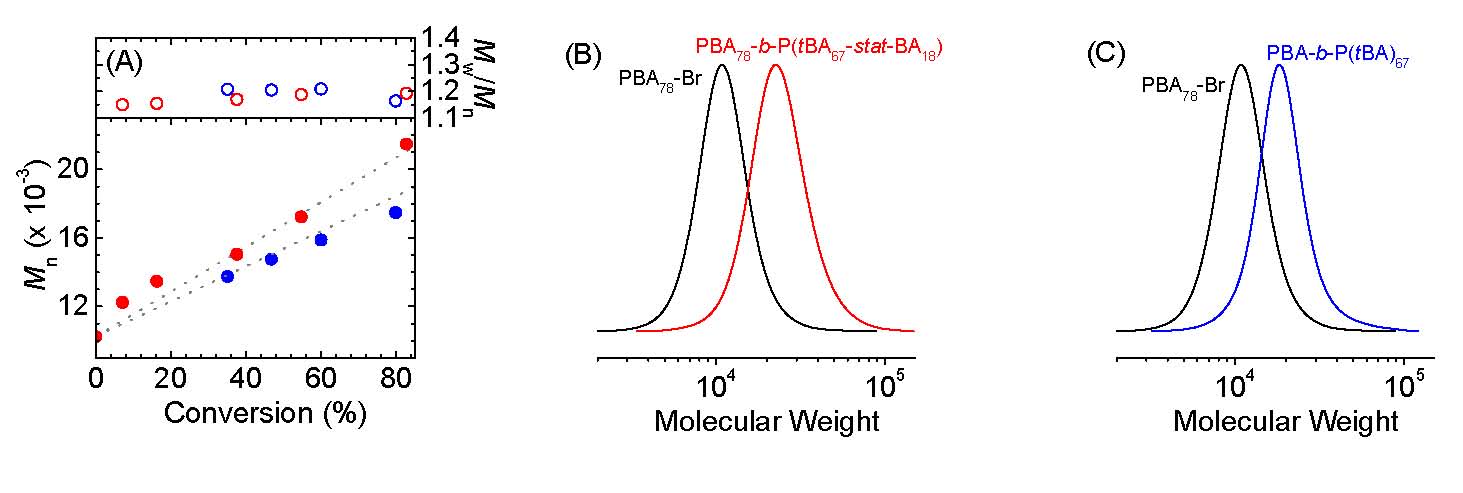
These results show that miniemulsion eATRP can be carried out with an anionic surfactant and a single, strongly hydrophilic catalyst. The obtained PBA was well-defined, with MWs matching theoretical values and low Đ. Chain extension experiments confirmed that chain-end functionality was very well retained. These results are in contrast with a previous paradigm that taught that superhydrophobic catalysts are required for ATRP in dispersed media.
In this work, the catalytic system was generated in situ by mixing commercially available reagents (NaBr, SDS, traditional copper catalysts). Only a few ppm of catalyst were present inside the monomer droplets. Polymer purification was simplified because, after crashing the miniemulsion, >99% of the hydrophilic catalyst was present in the aqueous phase in contrast with a previous paradigm that superhydrophobic catalysts are required for ATRP in dispersed media. Indeed starting with 50 ppm catalyst in the polymerization resulted in less than 300 ppb catalyst in the final product. (28)
The ideal Cu catalyst for harnessing the interaction between surfactant and hydrophilic ligand complex has the following characteristics: (i) high activity, (ii) ability to form ion pairs with SDS, and (iii) stronger affinity for the surfactant when present in its CuII oxidation state.
Further studies set out to look for suitable ligands and examine the role of hydrophilicity and ligand activity while seeking to develop a low ppm Cu system that will allow simplification of experimental setup in an eATRP: e.g. aluminum wire anode, and/or a non-Pt cathode, and whether the surfactant assisted formation of a catalyst for eATRP miniemulsion polymerization could be extended to other low catalyst reactivation procedures such as ARGET (28) which is discussed in the “Surfactant Interaction with a Hydrophilic Catalyst for a Miniemulsion Polymerization” section of the “Heterogeneous Polymerization Media” segment in the general discussion on conducting an “ATRP in Protic Media”.
REFERENCES
(1) Matyjaszewski, K.; Jakubowski, W.; Min, K.; Tang, W.; Huang, J.; Braunecker, W. A.; Tsarevsky, N. V. Proceedings of the National Academy of Sciences 2006, 103, 15309-15314.(2) Min, K.; Gao, H.; Matyjaszewski, K. J. Am. Chem. Soc. 2006, 128, 10521-10526.
(3) Qiu, J.; Matyjaszewski, K.; Thouin, L.; Amatore, C. Macromol. Chem. Phys. 2000, 201, 1625-1631.
(4) Matyjaszewski, K.; Xia, J. Chem. Rev. 2001, 101, 2921-2990.
(5) Tang, W.; Matyjaszewski, K. Macromolecules 2006, 39, 4953-4959.
(6) Braunecker, W. A.; Brown, W. C.; Morelli, B. C.; Tang, W.; Poli, R.; Matyjaszewski, K. Macromolecules 2007, 40, 8576-8585.
(7) Nasser-Eddine, M.; Delaite, C.; Dumas, P.; Vataj, R.; Louati, A. Macromolecular Materials and Engineering 2004, 289, 204-207.
(8) Bonometti, V.; Labbe, E.; Buriez, O.; Mussini, P.; Amatore, C. Journal of Electroanalytical Chemistry 2009, 633, 99-105.
(9) Magenau, A. J. D.; Strandwitz, N. C.; Gennaro, A.; Matyjaszewski, K. Science 2011, 332, 81-84.
(10) Matyjaszewski, K.; Dong, H.; Jakubowski, W.; Pietrasik, J.; Kusumo, A. Langmuir 2007, 23, 4528-4531.
(11) Pintauer, T.; Matyjaszewski, K. Chemical Society Reviews 2008, 37, 1087-1097.
(12) Matyjaszewski, K.; Bortolamei, N.; Magenau, A.; Gennaro, A.; Isse, A. A. In WIPO; (Carnegie Mellon University, USA). Application: WO 2012091965, 2012; p 69pp.
(13) Magenau, A. J. D.; Bortolamei, N.; Frick, E.; Park, S.; Gennaro, A.; Matyjaszewski, K. Macromolecules (Washington, DC, U. S.) 2013, 46, 4346-4353.
(14) Bortolamei, N.; Isse, A. A.; Magenau, A. J. D.; Gennaro, A.; Matyjaszewski, K. Angew. Chem., Int. Ed. 2011, 50, 11391-11394, S11391/11391-S11391/11315.
(15) Chmielarz, P.; Park, S.; Simakova, A.; Matyjaszewski, K. Polymer 2015, 60, 302-307.
(16) Hosseiny, S. S.; van Rijn, P. Polymers (Basel, Switzerland) 2013, 5, 1229-1240.
(17) Chmielarz, P.; Krys, P.; Park, S.; Matyjaszewski, K. Polymer 2015, 71, 143-147.
(18) Shida, N.; Koizumi, Y.; Nishiyama, H.; Tomita, I.; Inagi, S. Angew. Chem., Int. Ed. 2015, 54, 3922-3926.
(19) Tsarevsky, N. V.; Pintauer, T.; Matyjaszewski, K. Macromolecules 2004, 37, 9768-9778.
(20) Millard, P.-E.; Mougin, N. C.; Boeker, A.; Mueller, A. H. E. ACS Symp. Ser. 2009, 1023, 127-137.
(21) Zhang, Q.; Wilson, P.; Li, Z.; McHale, R.; Godfrey, J.; Anastasaki, A.; Waldron, C.; Haddleton David, M. J Am Chem Soc 2013, 135, 7355-7363.
(22) Park, S.; Chmielarz, P.; Gennaro, A.; Matyjaszewski, K. Angew. Chem., Int. Ed. 2015, 54, 2388-2392.
(23) Wang, Y.; Fantin, M.; Park, S.; Gottlieb, E.; Fu, L.; Matyjaszewski, K., Macromolecules 2017, 50, 7872-7879.
(24) Fantin, M.; Park, S.; Wang, Y.; Matyjaszewski, K., Macromolecules 2016, 49 (23), 8838-8847.
(25) Fantin, M.; Chmielarz, P.; Wang, Y.; Lorandi, F.; Isse, A. A.; Gennaro, A.; Matyjaszewski, K., Macromolecules 2017, 50 (9), 3726–3732.
(26) Gaynor, S. G.; Qiu, J.; Matyjaszewski, K., Macromolecules 1998, 31 (17), 5951-5954.
(27) Teo, V. L.; Davis, B. J.; Tsarevsky, N. V.; Zetterlund, P. B., Macromolecules 2014, 47 (18), 6230-6237.
(28) Wang, Y.; Lorandi, F.; Fantin, M.; Chmielarz, P.; Isse, A. A.; Gennaro, A.; Matyjaszewski, K., Macromolecules 2017, 50 (21), 8417-8425.