Iron Based ATRP
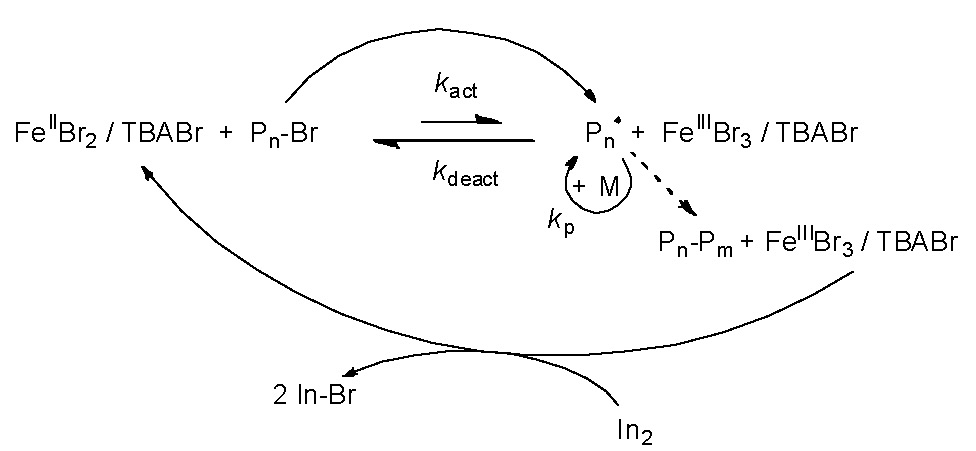
“Classic” Iron Based ATRP
Mechanistic Side Reactions
Novel aspects of Iron Based Catalysts
“Classic” Iron Based ATRP
Iron is the most abundant transition metal element in the earth’s crust, it has been employed as a catalyst for many organic reactions and is environmentally friendly (non toxic). Iron based ATRP catalyst systems go back to the initial use of zero valent metals, in conjunction with a suitable ligand, as precursors of catalyst systems for atom transfer radical polymerization in 1997.(1-2) The initial paper described the use of zero valent metals themselves or in conjunction with other, higher, oxidation state metal salts, i.e., copper(0)/copper(I), copper(0)/copper(II), iron(0)/iron(II), and iron(0)/iron(III) as suitable catalyst precursors. It was noted that the addition of these simple metals to the soluble catalyst system (e.g. copper(I), iron(II)/Ligand) provided an increase in the rate of polymerization while still maintaining good control over the polymerization of styrenes, and (meth)acrylates. A paper submitter earlier (3) described the use of iron halide complexes under both homogeneous and heterogeneous conditions for the ATRP of styrene and methyl methacrylate with a variety of coordinating ligands including 4,4'-bis(5-nonyl)-2,2'-bipyridine, trialkylamines, triphenylphosphine, trialkylphosphines, and trialkylphosphites.
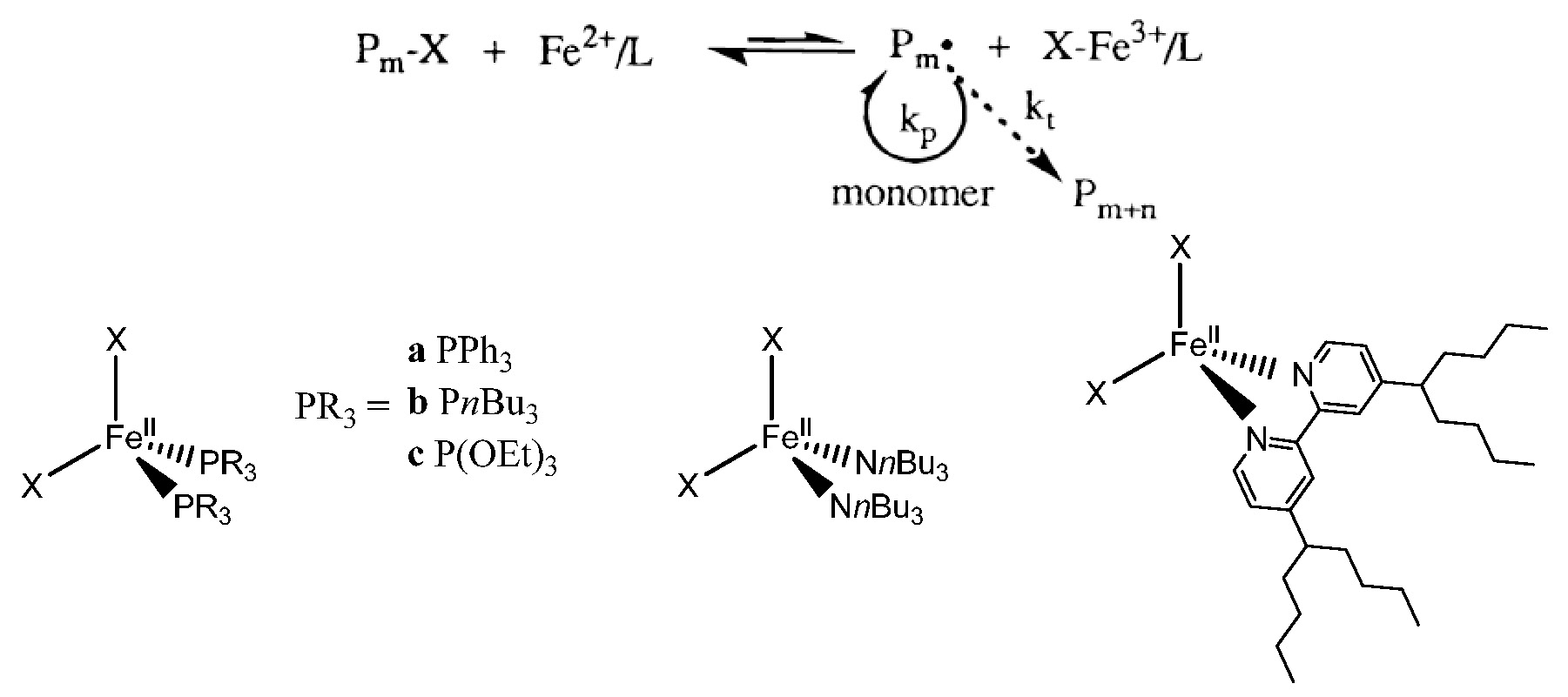
Both the polymerization rate and molecular weight distribution (Mw/Mn = 1.1-1.5) were influenced by the structure of the coordinating ligands and the monomers employed. Sawamoto also reported ironIIbis(triphenylphosphine)dichloride [FeCl2(PPh3)2] induced living radical polymerization of methyl methacrylate (MMA) in 1997 (4) and noted that, unlike the initial RuCl2(PPh3)3 counterpart, (5) the reaction did not require the presence of an aluminum based activator.
Subsequently, a simplification of the iron based ATRP catalyst systems was reported.(6) The direct use of iron(II) bromide complexes formed in the presence of ammonium and phosphonium chloride, bromide, or iodide salts as complexing ligands were shown to catalyze the polymerization of both St and (meth)acrylates in a controlled manner. The experimental molecular weights increased linearly with monomer conversion and were close to the theoretical values. The polymerization rates and molecular weight distributions (Mw/Mn = 1.1-1.4) were dependent on the monomer employed.
Reverse ATRP, initiated by AIBN in the presence of FeBr3/onium salts, provided a controlled polymerization of both MMA and methyl acrylate (MA), but the involvement of cationic polymerization in the St polymerization procedure led to the formation of polymers with uncontrolled molecular weight and high Mw/Mn. The most frequently used iron based catalysts were FeIIBr2/FeIIIBr3 or FeIICl2/FeIIICl3 with amine, imine, or phosphine ligands.
Some ligands used for iron based ATRP are shown in the following figure and their utility is discussed in a recent review paper.(7)
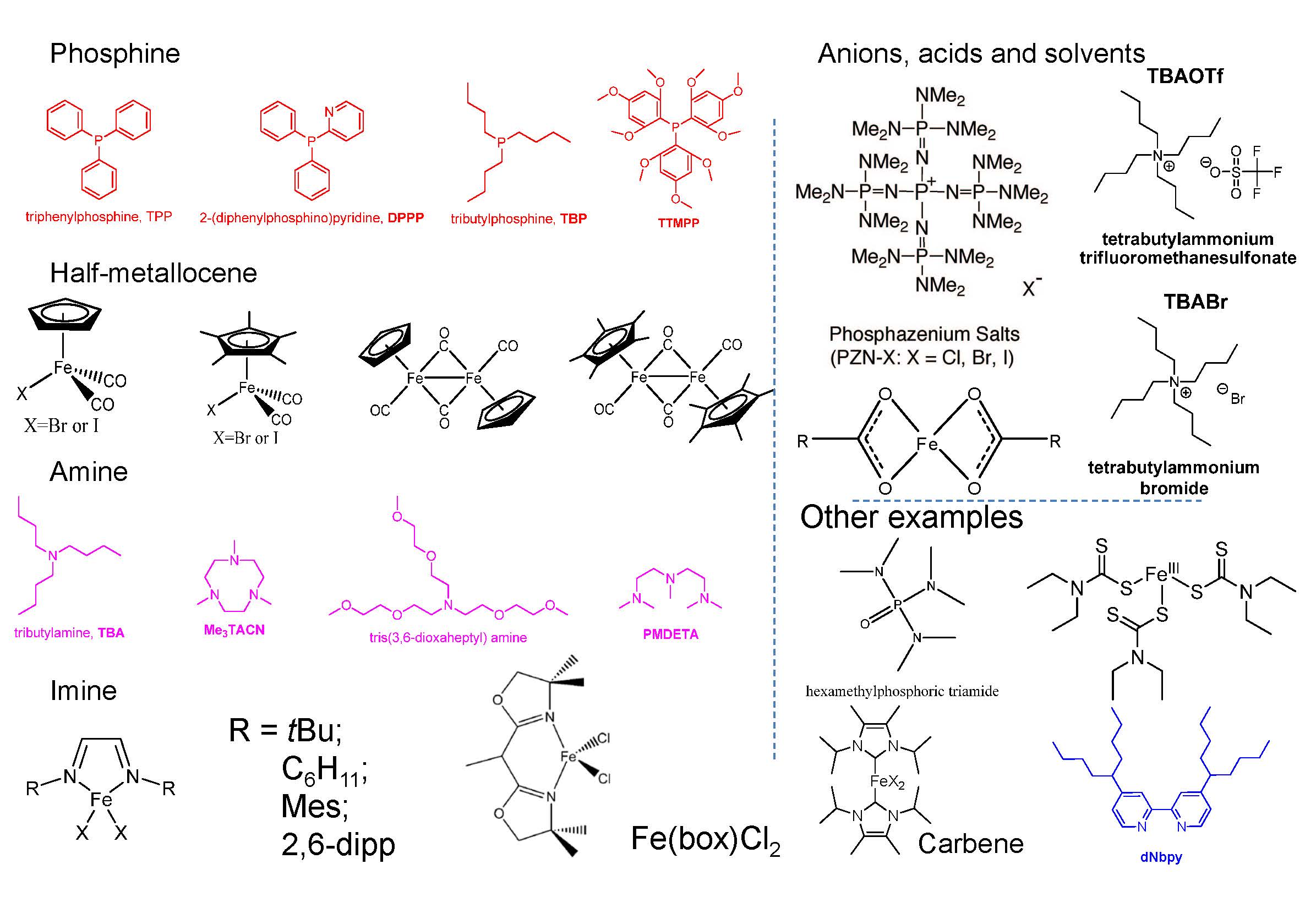
Grubbs examined the use of the imidazolylidene ligands shown below, with both chlorine and bromine ions, and demonstrated that they formed highly reactive and efficient catalysts for the ATRP of St and MMA with PEBr at 85 oC, and EBiB at 60 oC, respectively.(8) The high activity was attributed to the high electron donacity of the ligand. Apart from being less toxic than phosphines and amines, imidazolylidenes are attractive because they are easy to prepare and handle.
Mechanistic Side Reactions
Iron based catalysts can participate in multiple polymerization processes including ATRP, organometallic radical polymerization (OMRP), catalytic chain transfer (CCT), and cationic polymerization procedures.
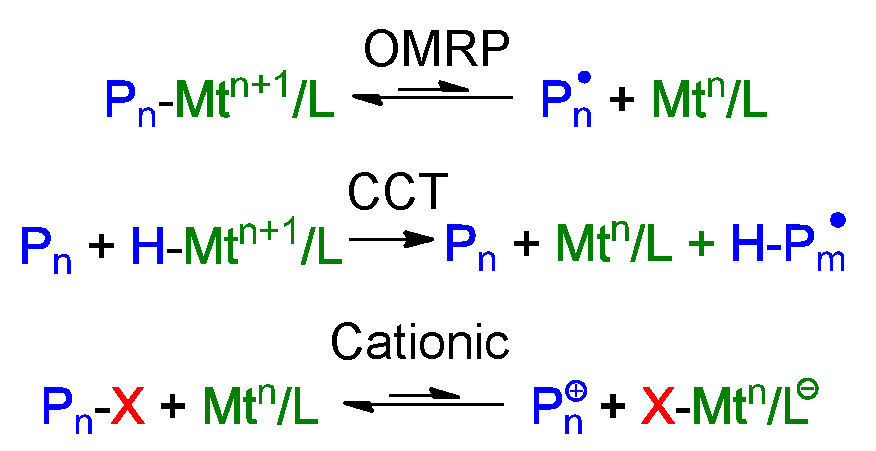
This was discussed in detail by Rinaldo Poli (9) and the effect of spin state of the metal in the iron complexes was discussed by Shaver and Gibson (10, 11) who noted that high-spin catalysts are halogenophilic, resulting in ATRP, while intermediate-spin complexes are carbophilic giving rise to CCT.
When the effect of the electron donating ability of substituents in α-diimine iron complex was examined it was determined that electron-donating substituents favor ATRP while electron-withdrawing substituents favor CCT. Furthermore, an analysis of the radical concentrations generated by the competing OMRP and ATRP equilibria indicates that the halogenophilicity of the FeII catalyst dominates the carbophilic alkyl radical-trapping capacity of the FeII species in this R-diimine catalyst system. In summary a catalyst complex forming a high spin is favored to avoid OMRP and CCT; addition of a Lewis base is needed to shut down cationic polymerization when St is the targeted monomer. Ligands forming catalysts with higher reducing power gives higher activity, halide affinity should also be considered and larger kdeact values provide better control.
Novel Aspects of Iron Based Catalysts
The ionic nature of the iron complexes means that they can be removed easily from the reaction mixture by washing with water. This simplified purification procedure was also reported for iron mediated ATRP carried out in the presence of ionic liquids.(12) The ionic liquids containing transition metal salts could be readily separated from the polymerization media and regeneration of the catalyst in the ionic liquid was successful.
A successful approach to more active iron based catalysts was reported by O’Reilly and Gibson when they discussed the preparation and use of a series of five-coordinate complexes of FeII containing tridentate nitrogen donor ligands for the ATRP of St.(13) Cyclic voltammetric studies showed that the redox potential for the FeII/FeIII couple strongly depended on the donor capacity of the complexing ligand. The polymerizations revealed that ligands derived from alkyl amine or pyridine groups were the most active although slower than their four-coordinate relatives due to unfavorable steric interactions. In general, catalyst activity decreases in the order of donor group: alkyl amine ~ pyridine > alkyl imine ≫aryl imine > aryl amine, with the aryl derivatives being almost completely inactive in ATRP.(14) The trend in activity correlated with the redox potential of the corresponding complexes. Indeed α-diimines are excellent ligands for four-coordinate iron ATRP catalysts due to their ease of preparation and their amenability to modification.(15) The activity is related to reducing power with higher reducing power providing more active catalysts.
Activators generated by electron transfer, AGET ATRP, was extended to iron mediated ATRP when an oxidatively stable FeCl3/triphenylphosphine catalyst complex was used with ascorbic acid as the reducing agent for the polymerization of methyl methacrylate.(15) The polymerization could be successfully conducted in the presence of a limited amount of air and displayed the features of "living"/controlled free-radical polymerizations such as Mn increasing linearly with monomer conversion and generating polymers with narrow molecular weight distributions. The “living” feature of the obtained polymer was further verified by chain extension. AGET ATRP was also used for MMA polymerization using a novel catalyst system based on FeCl3 complexes with iminodiacetic acid and using ascorbic acid as a reducing agent. The kinetics of AGET ATRPs of MMA with different amounts of reducing agent in the presence of air was investigated.(16) An AGET ATRP of OEOMA475 was activated by addition of Sn(EH)2 to the ferric complex as a reducing agent and the polymerization displayed close to linear first order kinetic plots and linear MW evolution with conversion. Dispersities stayed low throughout course of polymerization.
In summary a catalyst complex forming a high spin is favored to avoid OMRP and CCT; addition of a Lewis base is needed to shut down cationic polymerization of styrene. Ligands forming catalysts with higher reducing power gives higher activity, halide affinity should also be considered and larger kdeact values provide better control.
ATRP of MMA has also been reported to occur in the presence of polar solvents where the reaction was catalyzed by FeBr2 without the need for additional ligand.(17, 18) The amount NMP could be reduced to as low as 10%, which still dissolved most of the FeBr2 and maintained control over the polymerization. The addition of a small amount of FeIIBr2 further improved control, as measured by a narrowing of the molecular weight distribution. The polymerization of MMA was conducted in non-polar solvents in the presence of a small amount of triflate anions. The weakly coordinating triflate anions helped dissolve FeIIBr2 in anisole but had little effect on the catalytic reactivity of the iron species. The ratio of triflate ions to FeII was varied from 0.5:1 to 2:1 and all reactions generated linear kinetics with narrow Mw/Mn = 1.13.(18)
The activation–deactivation equilibrium associated with iron-mediated ATRP under increasing levels of pressure was examined for FeBr2 activation of an α-bromoester model system in the absence of monomer.(19) Quantitative analysis was carried out in solution of either NMP or acetonitrile (MeCN) via high-pressure online VIS/NIR spectroscopy monitored the formation of the FeIII species. In contrast to the results determined for copper based catalyst complexes the equilibrium constant decreased with pressure, with a positive reaction volume of about Δ V R = 13 cm3 mol−1. The unexpected result of determining a positive reaction volume suggests that several iron–solvent species are present in the solution. Among them, the catalytically inactive [FeII(NMP)6]2+ species, is favored by applying pressure. A series of experiments examining iron-mediated ATRP of MMA suggested that the unusual pressure effect also occurs under normal polymerization conditions, that is, in the presence of monomer. The impact of pressure on the catalytic iron species and on the activation–deactivation equilibrium therefore appears to be adequately reflected by the model systems studied in the paper. The initial communication concluded that the solvent needs to be of sufficient polarity to ensure good solubility but, complexation should not be too strong to disfavor the formation of octahedral cationic FeII–solvent species. This study was confirmed in a later paper (20) where it was noted that the activation–deactivation equilibrium constant, KATRP, decreased in the presence of higher NMP content due to the formation of a catalytically less active FeII/NMP species. Furthermore it was observed that conducting an iron-catalyzed ATRP of MMA in less polar solvents, or even without the addition of a solvent (i.e., for bulk ATRP) yielded KATRP values, which were two to three orders of magnitude higher than the values obtained in the presence of NMP and, in further contrast to the observations made in the presence of NMP, high pressure increased the polymerization rate due to both an increase in kp and a lowering of the FeIII persistent radical concentration without unfavorably affecting dispersity. Conducting an iron-mediated ATRP in anisole solution may be a good compromise.(18)
As mentioned above, and in many of the cited papers, iron-mediated ATRP has been widely investigated because of its low toxicity and biocompatibility which is especially advantageous when targeting biological applications. ATRP in aqueous media provides several advantages. Water is an environmentally benign solvent, enabling direct polymerization of water-soluble monomers, providing faster reactions, and allowing polymerization in the presence of biomolecules.(21) In light of work conducted by di Lena (22) and Bruns (23) on metalloenzymes, such as heme proteins horseradish peroxidase, these results prompted research into a series of bio-inspired iron porphyrin-based complexes for ATRP. Rather than examine proteins directly as catalysts for ATRP of water soluble monomers synthetic analogues of the core of the functional bioinspired catalysts, such as horseradish peroxidase, catalase or hemoglobin were synthesized to overcome limitations observed for ATRP in the earlier papers.(24) Hemin, a ferric form of heme with a chloride ligand instead of hydroxyl group as in hematin was initially selected, however, this catalyst has low halidophilicity, low solubility in water, and can itself (co)polymerize because of the presence of vinyl moieties. As shown below the double bonds were hydrogenated and the acidic groups esterified with poly(ethylene glycol) methyl ether to provide a saturated metal complex with improved hydrophylicity.
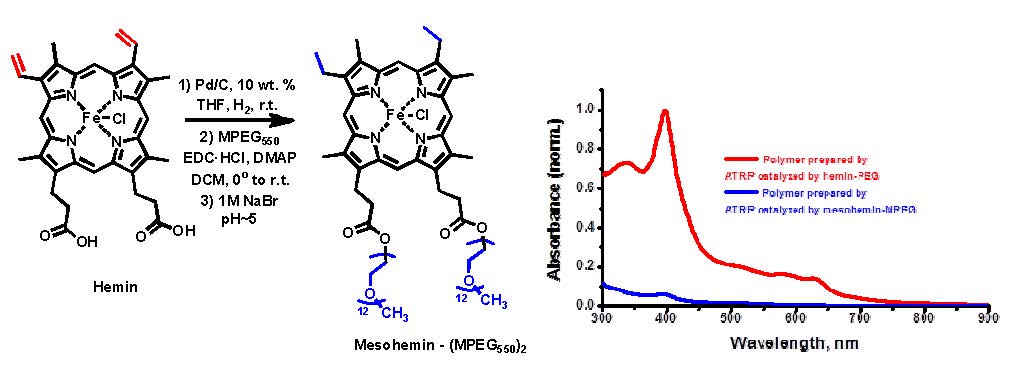
The novel hydrophilic mesohemin-(PEG550)2 complex performed significantly better than the precursor hemin as a catalyst for the ATRP of OEO(M)A475 in aqueous media in the presence of potassium bromide, to improve both rate of deactivation and initiation efficiency, a with different ratios of ascorbic acid as the reducing agent. The reactions were relatively fast, 60% conversion in 6 hours, providing linear first order kinetic plots, linear evolution of molecular weight and Mw/Mn values close to 1.20 with low ratio, 1:1, of ascorbic acid to initiator and catalyst. The mesohemin-(PEG550)2 complex also performed relatively well in organic media (anisole). Research is continuing with the target of increasing to activity of hemin derived catalyst complexes.
Increasing the Activity of Iron Based Catalysts
In 2008 it was reported that FeIIIX3 complexes formed with the various phosphines ligands, could mediate an ATRP in the absence of any radical initiator or additional reducing agent.(25, 26) The proposed mechanism was that the FeII species could be generated in-situ via a reaction between the FeIII complex and the monomer, illustrated in scheme (1), as previously reported for a CuIIX2/L system. (27) However, FeIIIBr3 could be also reduced in the presence of phosphine to generate a FeIIBr2 and dibromophosphorane, scheme (2).
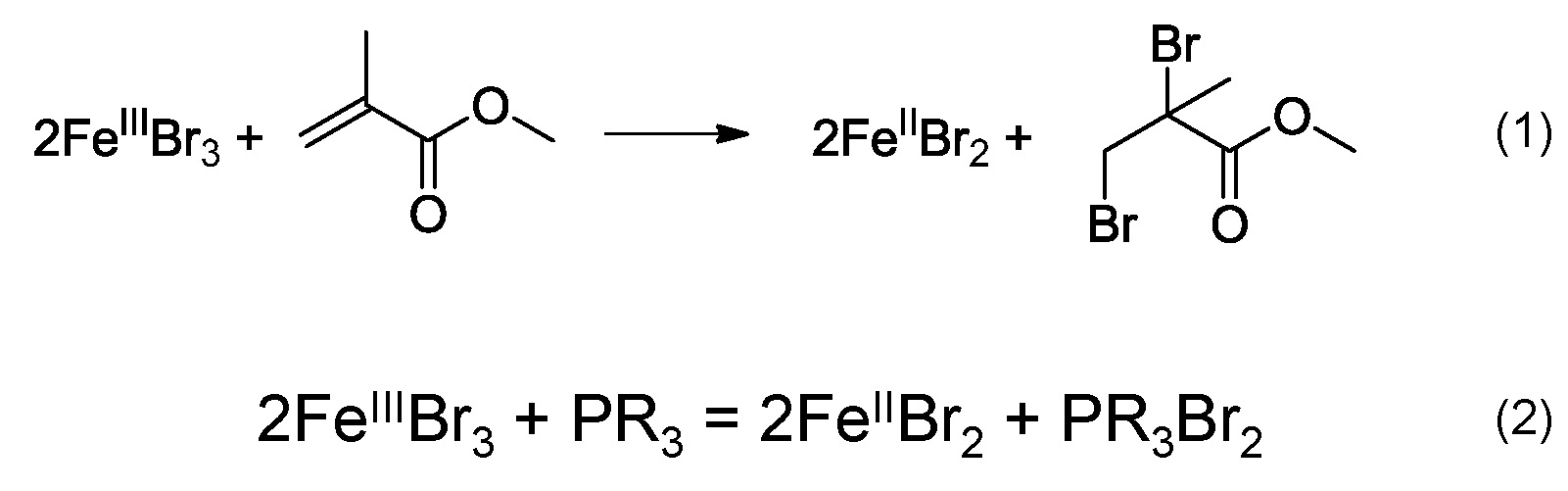
The substituents present on the phosphine ligands have a significant influence on the activity of the transition metal catalyst complexes. Higher activity means a larger value for KATRP, which is generally correlated with a faster polymerization under identical conditions. However, since phosphine can reduce FeIII to FeII, the faster rate of polymerization could be a result of both a larger value for KATRP and faster reduction of FeIII. Within the group it was recently determined that iron based ATRP of styrene with various triarylphosphines, containing electron donating methoxy groups, led to different rates of polymerization.(28) In order to determine whether phosphines with more electron donating groups on the aromatic rings will accelerate an ATRP three different phosphines: triphenylphosphine (TPP), tris(4-methoxyphenyl)phosphine (TMPP) and tris(2,4,6- trimethoxyphenyl)phosphine (TTMPP) were employed with FeIIIBr3 to control the polymerization of styrene.
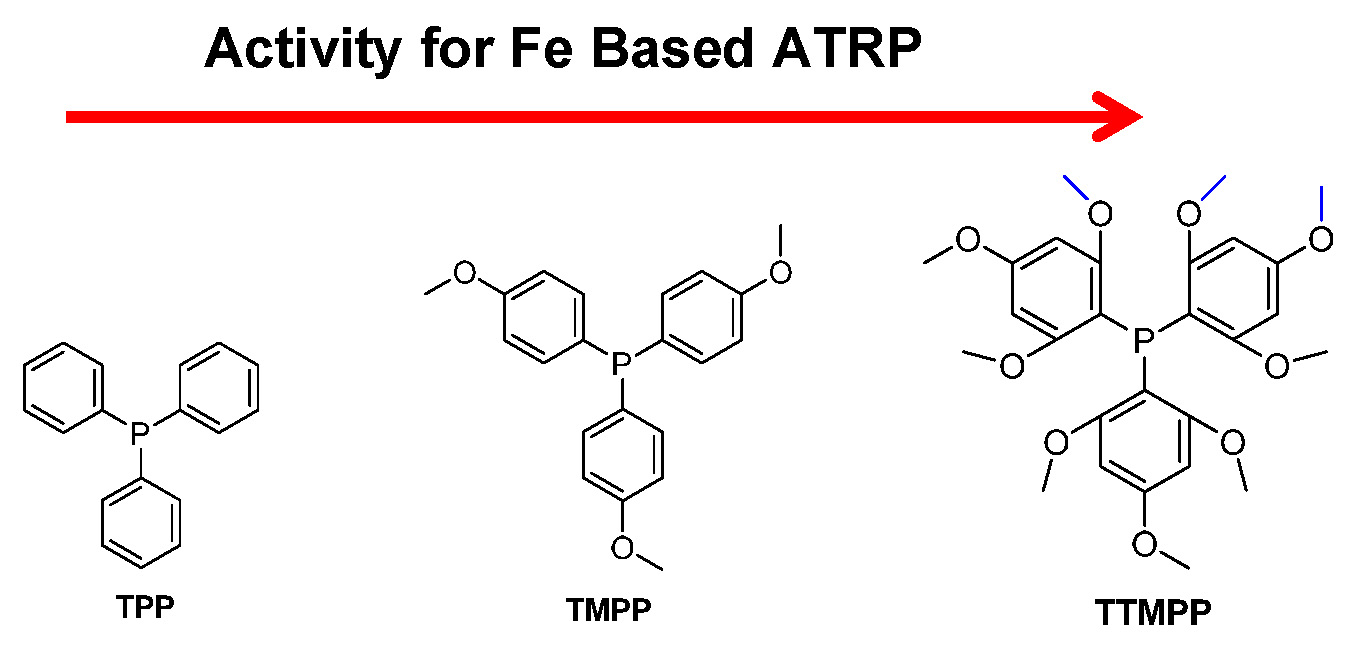
The ATRP of styrene with an FeIIIBr3 complex formed with the TTMPP ligand led to a faster polymerization than one formed with TMPP, which was much faster than the catalyst complex formed with TPP as ligand under identical conditions. The polymerizations were carried out with the initial ratio of reagents equal to [St]:[EBiB]:[FeIIIBr3]:[Ligand] = 200:1:1:2, in 50% (v/v) anisole at 100 oC, (EBiB is ethyl 2-bromoisobutyrate). Conversion reached 92% after 21 h in the presence of the FeIIIBr3/TTMPP catalyst complex yielding polystyrene with molecular weight Mn = 24,100 and Mw/Mn = 1.25. Whereas with TMPP or TPP as ligands, under the same conditions, conversion of monomer was only 19% and 9%, respectively. Control was better with one equivalent of TTMPP vs. FeIIIBr3, resulting in formation of a polymer with Mw/Mn ~ 1.1, but the rate of polymerization was slower indicating that the phosphines could directly reduce FeIII to FeII, and the FeII species acted as the actual activator. Therefore the FeIIIBr3/phosphine catalyzed ATRP can be considered as an activator generated by electron transfer (AGET) ATRP process, in which the phosphine acts as the reducing agent. A summary of the results is provided in the following table.
Ligand : Ratio to FeIII Mn,GPC Mn,th Conv. Mw/Mn
TPP :1 900 900 4% 1.05
TMPP : 1 1,300 2,400 12% 1.10
TTMPP :1 8,100 9,200 44% 1.11
TPP :2 1,200 1,800 9% 1.09
TMPP : 2 1,900 2,400 19% 1.15
TTMPP :2 24,100 19,100 92% 1.25
[St]:[EBiB]:[FeBr3]:[Ligand] = 200:1:1: (1 or 2), in 50%(v/v) anisole at 100 oC, after 21 h
The proof that phosphine can reduce FeIII to FeII was provided by the following set of experiments. A PSt-Br macroinitiator with Mn = 12,000 and Mw/Mn = 1.10 was prepared via normal ATRP. A reaction mixture with 0.1 g of this PSt-Br macroinitiator in 2 mL anisole was heated at 110 oC. The molecular weight and Mw/Mn value remained unchanged after 18 h. However, when 0.1 g PSt-Br was mixed with a catalyst complex with the ratio of reagents [PSt-Br]:[FeIIIBr3]:[TTMPP] = 1:1:2 in 2 mL anisole and heated at 110 oC for 18 h, a bimodal GPC curve was observed. The peak molecular weight increased from 14,000 to 26,800 as shown in the central GPC trace in the following figure. This observation suggested that PSt-Br could be activated by the FeII species forming a radical and the resulting macroradicals terminated via coupling, forming PSt with double the molecular weight.
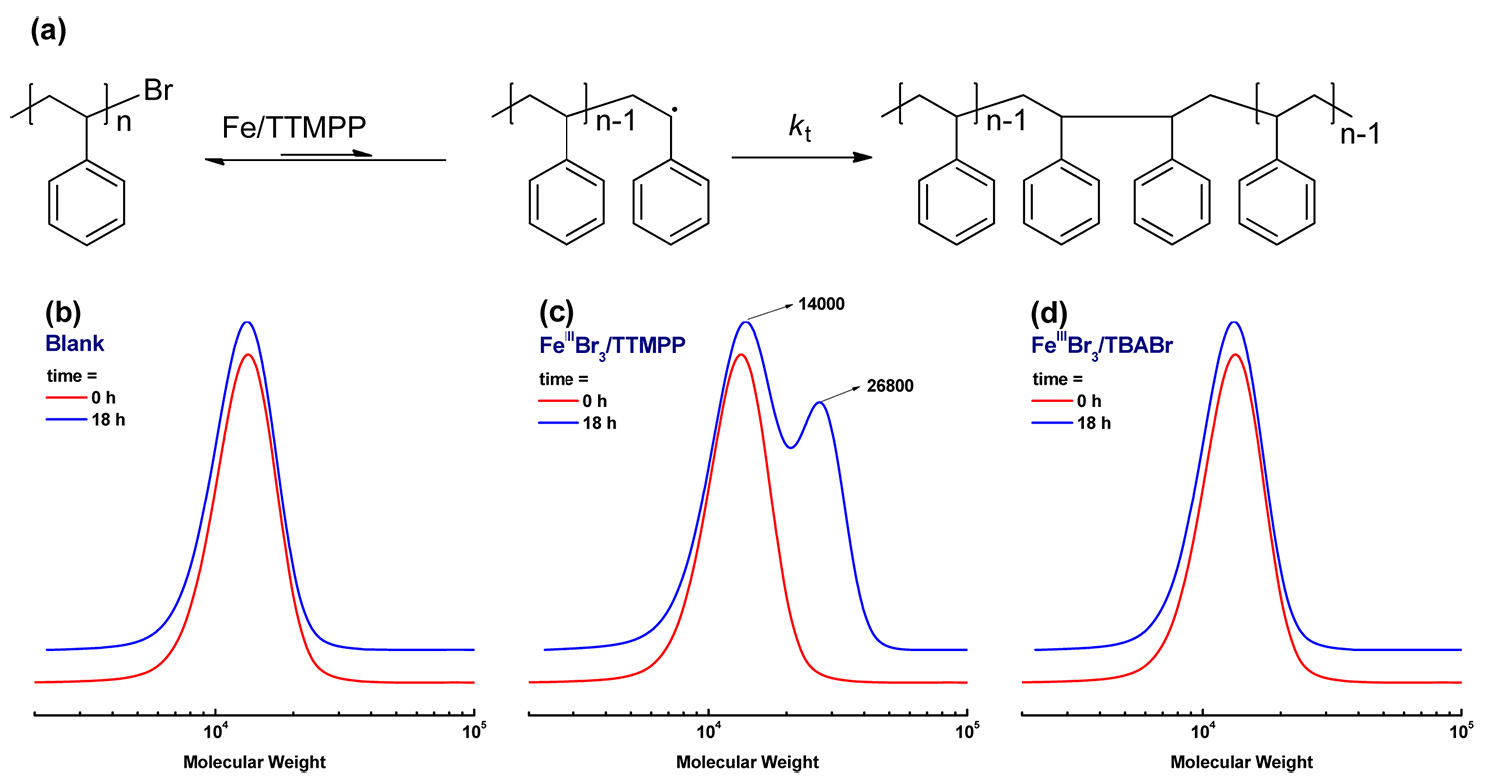
Indeed since there was no additional reducing agent or monomer in this reaction mixture, it is clear that TTMPP reduced FeIII to FeII which activated the dormant species. For comparison, the same experiment was performed with tetrabutylammonium bromide, TBABr, which was previously used as a successful co-catalyst for Fe based ATRP of St, instead of TTMPP. In this case, the macroinitiator could not be activated and the molecular weight did not change, even after 18 h, right hand GPC trace. These experiments suggested that in Fe based ATRP processes phosphines can act as reducing agents.
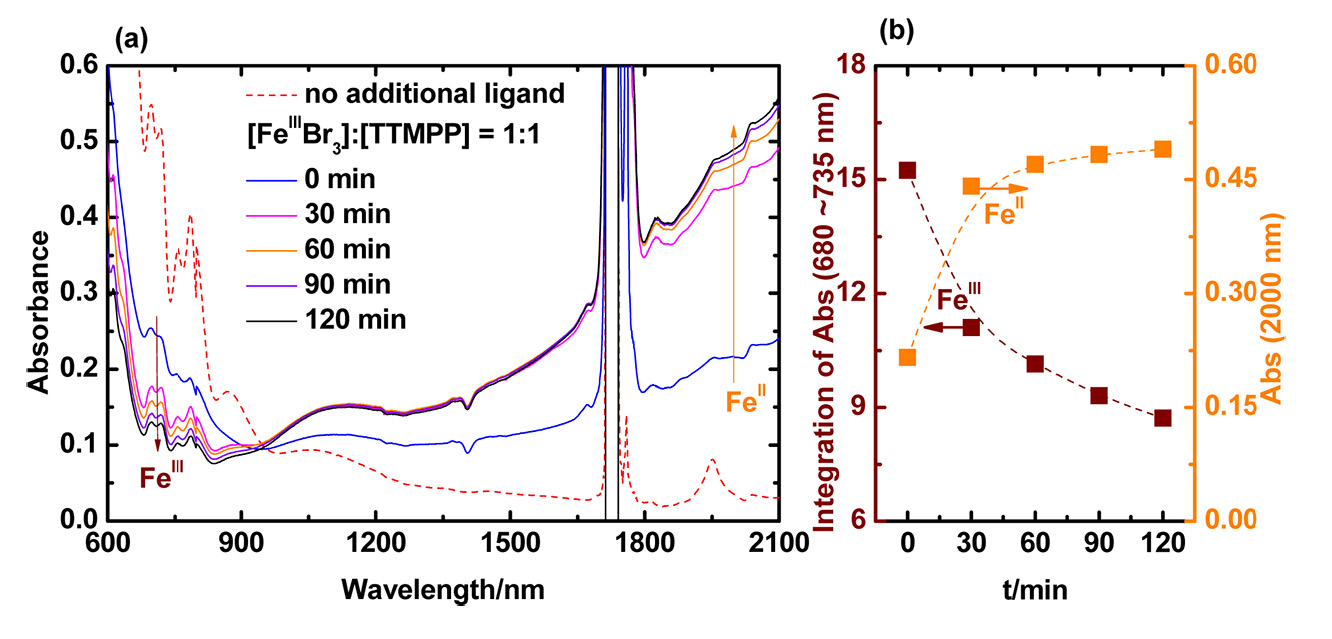
The reduction process was studied via UV/VIS/NIR spectroscopy of 30 mM FeIIIBr3 in DMF in the presence of 1 equivalent of TTMPP at room temperature (23 oC) and the change of absorbance corresponding to FeIII and FeII was followed over time. The absorption bands of FeIII with halide ligands between 680 and 735 nm decreased strongly after addition of 1 equiv of TTMPP. The absorbance continued to decrease over 2 h, indicating a continuous gradual reduction process. The broad absorbance of FeII, which could be observed at 2000 nm, appeared quickly after addition of TTMPP and continued to increase during 2 h. The reduction of FeIII by phosphine should result in the corresponding dibromophosphorane.
A successful ICAR ATRP of styrene was conducted with iron(III) bromide and 1,1'-azobis(cyclohexanecarbonitrile) (ACHN) as the thermal initiator. The polymerization was started with 50 ppm of FeBr3 and 50 mol equiv. of ACHN in 33% (vol./vol.) anisole at 90 °C, reached 70% conversion in 24 h and was well controlled, giving a polymer with a narrow MWD, Mw/Mn = 1.15. which corresponded well to theoretical values, as conversion increased.(29)
A polystyrene with a relatively narrow molecular weight distribution, Mw/Mn = 1.29 at 65% of conversion, was obtained with 5 ppm of FeBr3 and the appropriate amount of ACHN.(30) This procedure therefore provides an efficient controlled polymerization in addition to creating a robust, cheap, and environmentally friendly catalytic system. The level of control over the polymerization with ACHN as the radical initiator in the ICAR ATRP was better than with tert-butyl peracetate as a thermal initiator or tinII 2-ethylhexanoate, Fe0, or Zn0 wire as reducing agents.
Therefore AGET, SARA and ICAR ATRP all work with iron based catalysts.
Molecules with complex topology can also be prepared using iron based catalyst systems. An FeBr2,/FeBr3, tetrabutylammonium bromide catalyst was employed for an ATRP dense “grafting-from” reaction of styrene, butyl acrylate and diethylene glycol ethyl acrylate resulting in formation of brush macromolecules with a narrow molecular weight distribution (Mw/Mn = 1.18-1.28).(31) Initiation efficiencies were calculated by cleaving the side chains by alcoholysis and then injecting to product into a gel permeation chromatography column. The initiation efficiencies were ca. 80-95 %, showing relatively higher values for the “grafting from” polymerization with an iron catalyst than with copper based systems. Furthermore the rate of polymerization with iron based catalysts was more sensitive to temperature than observed with copper based catalyst systems. The brush macromolecule formed with PDEGA side chains displayed theromoresponsive properties, a LCST, in aqueous solution. These results indicate that iron catalyzed ATRP can provide well controlled polymerizations even when targeting dense grafting from procedures.
REFERENCES
(1) Matyjaszewski, K.; Coca, S.; Gaynor, S. G.; Wei, M.; Woodworth, B. E. Macromolecules 1997, 30, 7348-7350.
(2) Matyjaszewski, K.; Gaynor, S. G.; Coca, S. In PCT Int. Appl.; (Carnegie Mellon University, USA). WO 9840415, 1998; p 230 pp.
(3) Matyjaszewski, K.; Wei, M.; Xia, J.; McDermott, N. E. Macromolecules 1997, 30, 8161-8164.
(4) Ando, T.; Kamigaito, M.; Sawamoto, M. Macromolecules 1997, 30, 4507-4510.
(5) Kato, M.; Kamigaito, M.; Sawamoto, M.; Higashimura, T. Macromolecules 1995, 28, 1721-1723.
(6) Teodorescu, M.; Gaynor, S. G.; Matyjaszewski, K. Macromolecules 2000, 33, 2335-2339.
(7) di Lena, F.; Matyjaszewski, K. Prog. Polym. Sci. 2010, 35, 959-1021.
(8) Louie, J.; Grubbs, R. H. Chemical Communications (Cambridge, United Kingdom) 2000, 1479-1480.
(9) Poli, R. Angewandte Chemie, International Edition 2006, 45, 5058-5070.
(10) Shaver, M. P.; Allan, L. E. N.; Gibson, V. C. Organometallics 2007, 26, 4725-4730.
(11) Shaver, M. P.; Allan, L. E. N.; Rzepa, H. S.; Gibson, V. C. Angewandte Chemie, International Edition 2006, 45, 1241-1244.
(12) Sarbu, T.; Matyjaszewski, K. Macromolecular Chemistry and Physics 2001, 202, 3379-3391.
(13) O'Reilly, R. K.; Gibson, V. C.; White, A. J. P.; Williams, D. J. J. Am. Chem. Soc. 2003, 125, 8450-8451.
(14) O'Reilly, R. K.; Gibson, V. C.; White, A. J. P.; Williams, D. J. Polyhedron 2004, 23, 2921-2928.
(15) O'Reilly, R. K.; Shaver, M. P.; Gibson, V. C.; White, A. J. P. Macromolecules 2007, 40, 7441-7452.
(16) Zhang, L.; Cheng, Z.; Shi, S.; Li, Q.; Zhu, X. Polymer 2008, 49, 3054-3059.
(17) Wang, Y.; Matyjaszewski, K. Macromolecules 2010, 43, 4003-4005.
(18) Wang, Y.; Matyjaszewski, K. Macromolecules 2011, 44, 1226-1228.
(19) Schroeder, H.; Yalalov, D.; Buback, M.; Matyjaszewski, K. Macromol. Chem. Phys. 2012, 213, 2019-2026.
(20) Schroeder, H.; Buback, M.; Matyjaszewski, K. Macromol. Chem. Phys. 2014, 215, 44-53.
(21) Averick, S.; Simakova, A.; Park, S.; Konkolewicz, D.; Magenau, A. J. D.; Mehl, R. A.; Matyjaszewski, K. ACS Macro Lett. 2012, 1, 6-10.
(22) Ng, Y.-H.; di Lena, F.; Chai, C. L. L. Chem. Commun. (Cambridge, U. K.) 2011, 47, 6464-6466.
(23) Kali, G.; Silva, T. B.; Sigg, S. J.; Seidi, F.; Renggli, K.; Bruns, N. ACS Symp. Ser. 2012, 1100, 171-181.
(24) Simakova, A.; Mackenzie, M.; Averick, S. E.; Park, S.; Matyjaszewski, K. Angew. Chem., Int. Ed. 2013, 52, 12148-12151.
(25) Xue, Z.; Nguyen, T. B. L.; Noh, S. K.; Lyoo, W. S. Angew. Chem., Int. Ed. 2008, 47, 6426-6429.
(26) Satoh, K.; Aoshima, H.; Kamigaito, M. J. Polym. Sci., Part A: Polym. Chem. 2008, 46, 6358-6363.
(27) Nanda, A. K.; Hong, S. C.; Matyjaszewski, K. Macromolecular Chemistry and Physics 2003, 204, 1151-1159.
(28) Wang, Y.; Kwak, Y.; Matyjaszewski, K. Macromolecules 2012, 45, 5911-5915.
(29) Wang, Y.; Zhang, Y.; Parker, B.; Matyjaszewski, K. Macromolecules 2011, 44, 4022-4025.
(30) Mukumoto, K.; Wang, Y.; Matyjaszewski, K. ACS Macro Lett. 2012, 1, 599-602.
(31) Mukumoto, K.; Li, Y.; Nese, A.; Sheiko, S. S.; Matyjaszewski, K. Macromolecules 2012, 45, 9243-9249.