Features of Controlled/"Living" Radical Polymerizations
This summary, which discusses the features of a CRP, is based on a review published in 2001.(1)
It is widely accepted that a controlled polymerization process should display the following features:
-
First-order Kinetic Behavior
-
Pre-determinable Degree of Polymerization
-
Designed (Usually) Narrow Molecular Weight Distribution
-
Long-lived Polymer Chain with Preserved End Functionalities
Introduction:
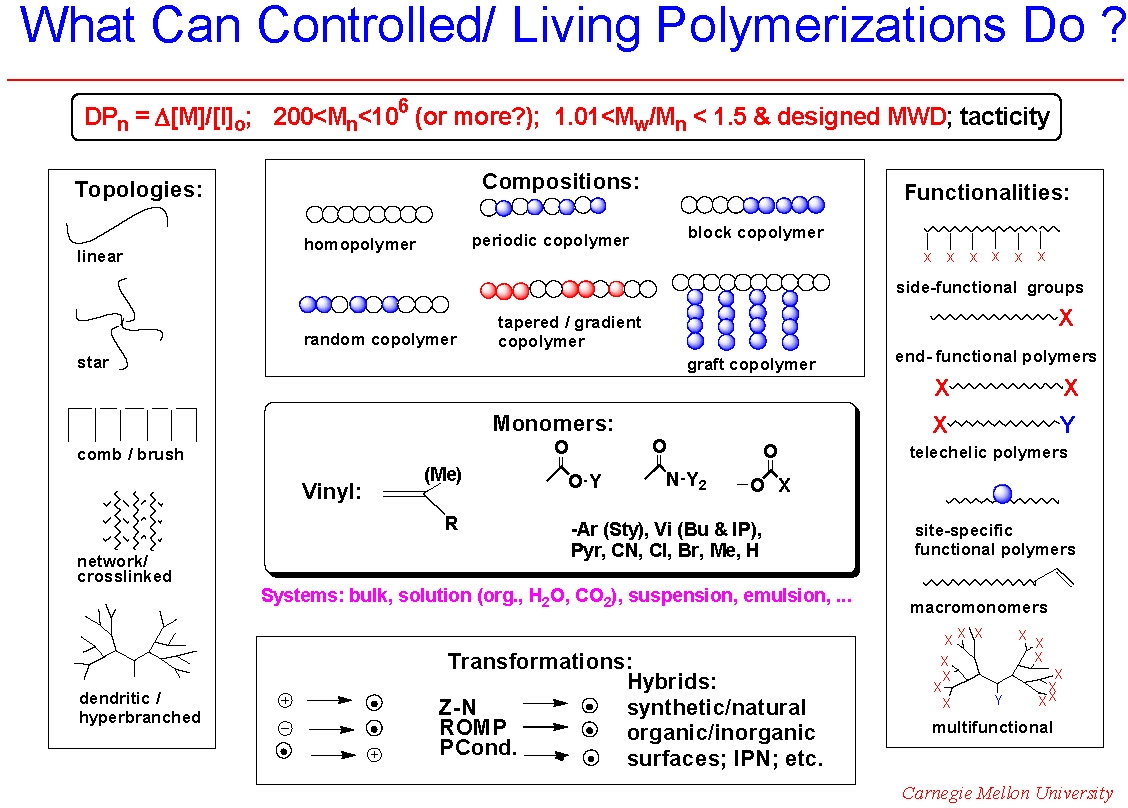
The introductory slide from the home page, reproduced below, indicates that Controlled/"Living" Radical Polymerization (CRP) procedures can be used for the preparation of copolymers, incorporating a broad spectrum of radically (co)polymerizable monomers forming materials with predetermined molecular weight, and narrow molecular weight distribution.(2) The most recent work on conducting an ATRP with low concentration of transition metals indicates that some control over the breadth of the MWD is also possible.(3) Here, and elsewhere in the text the word "control" and/or "controlled" means that if the polymerization process conditions are selected so that the contributions of the chain breaking processes are insignificant compared to chain propagation, then synthesis of polymers with predetermined molecular weights, low polydispersity and site specific functionalities become a reality.
FEATURE 1 First-order kinetic behavior:
i.e. the polymerization rate (Rp) with respect to the log of the monomer concentration ([M]) is a linear function of time. This is due to the negligible contribution of non-reversible termination, so that the concentration of the active propagating species ([P*]) is constant.
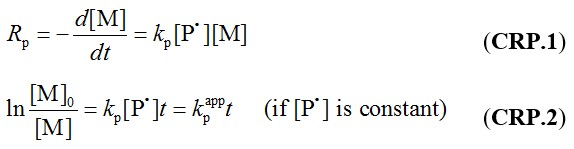
kp is the propagation constant.
The consequence of equation CRP.2 and the effect of changes in P* are illustrated in below:
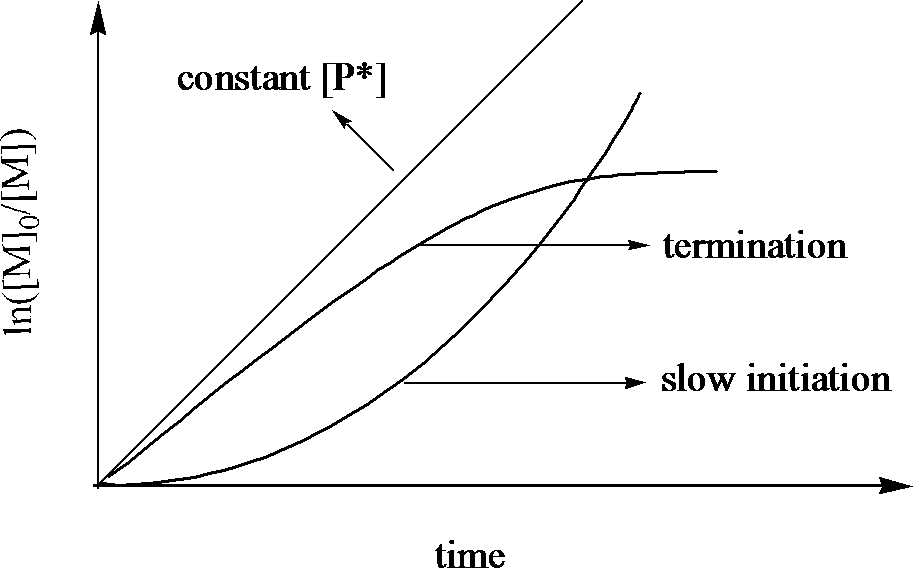
Illustration of FEATURE 1, the depence of Ln([M]0/[M]) on time
This semilogarithmic plot is very sensitive to any change of the concentration of the active propagating species. A constant [P*] is revealed by a straight line. A steady [P*] in a CRP is established by balancing the rates of activation and deactivation and not by balancing the rates of initiation and termination as in a conventional radical polymerization. An upward curvature in the kinetic plot indicates an increase in [P*], which occurs in case of slow initiation. On the other hand, a downward curvature suggests a decrease in [P*], which may result from termination reactions increasing the concentration of the persistent radical, or some other side reactions such as the catalytic system being poisoned or redox processes on the radical.It should also be noted that the semilogarithmic plot is not sensitive to chain transfer processes or slow exchange between different active species, since they do not affect the number of the active propagating species.
FEATURE 2 Pre-determinable degree of polymerization:
(DPn), i.e. the number average molecular weight (Mn) is a linear function of monomer conversion.
This result comes from maintaining a constant number of chains throughout the polymerization, which requires the following two conditions:
1) initiation should be sufficiently fast so that essentially all chains are propagating before the reaction is stopped; and
2) an absence of chain transfer reactions that increases the total number of chains.
The following figure illustrates feature 2 and shows the ideal growth of molecular weight with conversion, as well as the effects of slow initiation and chain transfer on the molecular weight evolution.
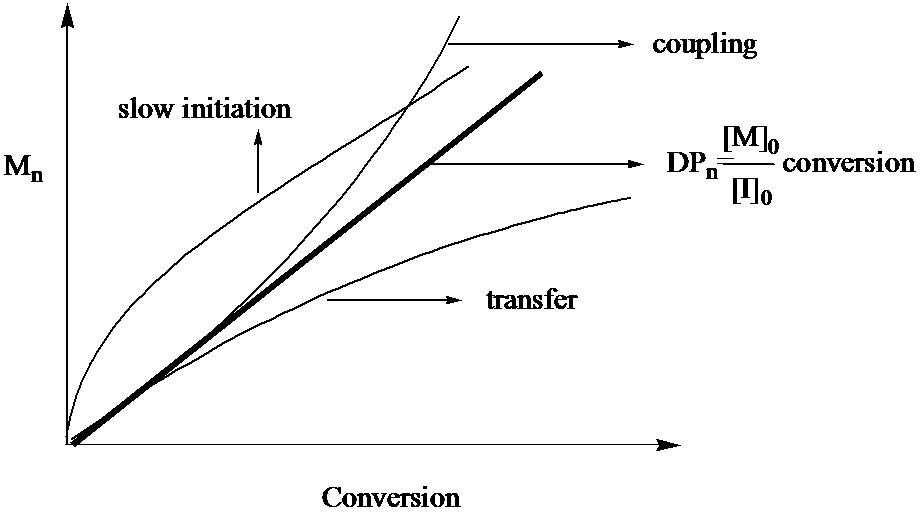
Figure illustrating FEATURE 2, the dependency of molecular weight on conversion
It is important to recognize that the evolution of molecular weight is not very sensitive to chain termination, since the number of chains remains unchanged. The effect of termination is only observable on the plot when coupling reactions, forming higher molecular weight polymers, start to play a significant role.
FEATURE 3 Designed, usually narrow, molecular weight distribution:
Although this feature is often desirable, it is not necessarily the result of a controlled polymerization, which only requires the absence of chain transfer and termination, but ignores the effect of rate of initiation, exchange and depropagation.Previous studies(4-6) indicate that in order to obtain a polymer with a narrow molecular weight distribution, each of the following requirements should be fulfilled.
i. The rate of initiation is fast in comparison to the rate of propagation. This condition allows simultaneous growth of all the polymer chains.
ii. The exchange between species of different reactivity is fast in comparison with the rate of propagation. This condition ensures that all the active chain termini are equally susceptible to reaction with monomer allowing uniform chain growth.
iii. There must be negligible chain transfer or termination.
iv. The rate of depropagation is substantially lower than propagation. This guarantees that polymerization is essentially irreversible.
This should yield a Poisson distribution, as quantified in equation CRP.4.
![]()
According to equation CRP.4, polydispersity (PDI = Mw/Mn) decreases with increasing molecular weight.
Systems with slow exchange do not follow this perfect distribution but PDI's in a well conducted ATRP are defined by the following equation, where kp is the rate of propagation, kdeact the rate of deactivation, [RX] the concentration of initiator and dormant chain ends and [CuIILnX] is the concentration of the higher oxidation state transition metal complex, the deactivator.(5)
![]()
A polymerization that satisfies the prerequisites listed above is expected to form a final polymer with a polydispersity less than 1.1 for Xn greater than 10. However, it is possible to fine tune PDI by varying [CuIILnX] or conversion. Wel-controlled segmented copolymers with higher PDI can self-organize to materials with interesting nanostructured morphologies.(3)
Feature 4. Long-lived polymer chains with preserved end-functionalities:
This is a consequence of negligible irreversible chain transfer and termination. Hence, all the chains retain their active centers after the full consumption of the monomer. Propagation resumes upon introduction of additional monomer. This unique feature enables the preparation of block copolymers by sequential monomer addition.
The significance of controlled polymerization as a synthetic tool is widely recognized and polymers having uniform predictable chain length are readily available. Controlled polymerization provides the best opportunity to control the bulk properties of a target material through control of the multitude of possible variations in composition, functionality and topology now attainable at a molecular level.
Through appropriate selection of the functional (macro)initiator, copolymers formed in a controlled/"living" polymerization process can have essentially any desired topology. Further, as noted at the foot of the figure showing what CRP can do, we highlight that mechanistic transformations permit the use of macroinitiators or macromonomers prepared by other polymerization procedures in many CRP processes which allows incorporation of a spectrum of functionalities and polymer segments prepared by any other controlled polymerization process into segments of copolymers prepared by CRP.
Indeed a plethora of previously unattainable polymeric materials have been prepared. Numerous examples of gradient,(5) block(6) and graft(7) copolymers have been reported, as well as polymers with complex architectures, including comb shaped polymer brushes,(8) stars,(9) and hyperbranched(10) copolymers. Progress has been made in the synthesis of each of these materials and the procedures are discussed in other sections of the web page.
REFERENCES
(1) Qiu, J.; Charleux, B.; Matyjaszewski, K. Polimery (Warsaw, Pol.) 2001, 46, 453-460.
(2) Matyjaszewski, K.; Xia, J. Chem. Rev. 2001, 101, 2921-2990.
(3) Listak, J.; Jakubowski, W.; Mueller, L.; Plichta, A.; Matyjaszewski, K.; Bockstaller, M. R. Macromolecules 2008, 41, 5919-5927.
(4) Coleman, B. D.; Fox, T. G. Journal of the American Chemical Society 1963, 85, 1241-1244.
(5) Matyjaszewski, K. Macromol. Symp. 1996, 111, 47-61.
(6) Matyjaszewski, K.; Editor. Cationic Polymerizations: Mechanisms, Synthesis, and Applications.; Dekker: New York, 1996.
(7) Matyjaszewski, K.; Ziegler, M. J.; Arehart, S. V.; Greszta, D.; Pakula, T. J. Phys. Org. Chem. 2000, 13, 775-786.
(8) Davis, K. A.; Matyjaszewski, K. Advances in Polymer Science 2002, 159, 2-166.
(9) Gaynor, S. G.; Matyjaszewski, K. ACS Symp. Ser. 1998, 685, 396.
(10) Beers, K. L.; Gaynor, S. G.; Matyjaszewski, K.; Sheiko, S. S.; Moeller, M. Macromolecules 1998, 31, 9413-9415.
(11) Sheiko, S. S.; Sumerlin, B. S.; Matyjaszewski, K. Prog. Polym. Sci. 2008, 33, 759-785.
(12) Matyjaszewski, K. Polymer International 2003, 52, 1559-1565.
(13) Gao, H.; Matyjaszewski, K. Prog. Polym. Sci. 2009, 34, 317-350.
(14) Matyjaszewski, K.; Gaynor, S. G. Macromolecules 1997, 30, 7042-7049.