Brush Macromolecules
SuperSoft Elastomers
Physical Crosslinking
Bond Scission
Some Other Properties of "Bottle Brush" Molecules
Ionic Conductors
Phototunable
Crystallizable
Stimuli Responsive Molecules
pH Responsive Materials
Dual Responsive Materials
Repeatable Fibrillar Adhesives
Brush Macromolecules
The major difference between graft copolymers discussed above and the following discussion on "bottle brush" macromolecules is the grafting density. In the vast majority of the papers written on bottle brush copolymers the graft density targeted, in at least one segment of the copolymer is one graft from each backbone monomer unit. The conformation and physical properties of the resulting brush-like macromolecules are controlled by the steric repulsion of the densely grafted side chains.(1)
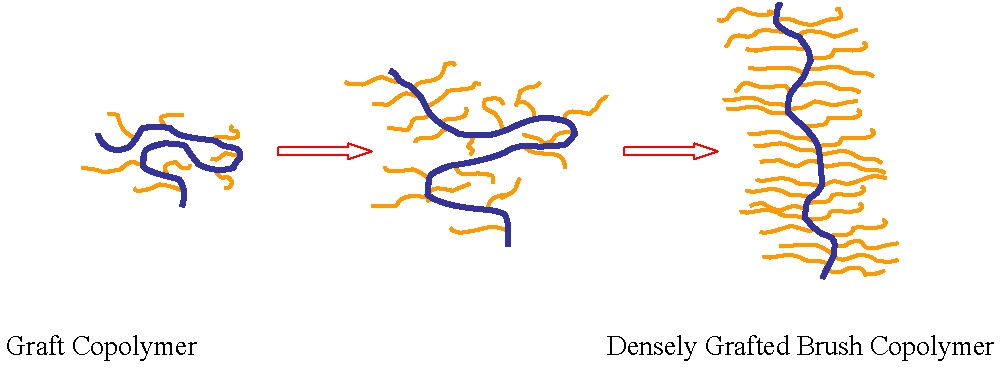
The result of such high graft density along the backbone is significant degree of side chain/side chain interactions, particularly close to the backbone of the copolymer, resulting in chain extension of the polymer backbone. As detailed below the molecules can be either flexible or rigid and can be designed to switch their conformation in response to changes in the surrounding environment; e.g., changes of temperature, solvent quality, pH, and ionic strength. Furthermore, one can control molecular conformation and related properties using external stimuli such as pressure, light and electro-magnetic fields.
The effect of graft density and side chain length on the structural mobility of a poly((2-(2-bromopropionyloxy)ethyl methacrylate-stat-methy methacrylate)-g-butyl acrylate) (poly((BPEM-stat-MMA)-g-PBA)) was examined by NMR relaxation dynamics.(2) The T2 values for protons of MMA units in the brush backbone significantly decreased with increasing side chain length and grafting density of PBA side chains. The mobility and relaxation times T2 for the side chain PBA protons displayed a more complex response. After an initial increase, the relaxation times eventually decreased with PBA side chain length indicating there was a difference in T2 values between the inner and outer protons of the side chains as the MW of the side chains increases. Both observations support the contention that there is molecular congestion along the backbone of a bottle-brush molecule but that congestion decreases as the MW of the tethered chain increases. However decoupling molecular mobility from differences in the chemical environment is difficult using 1H NMR. A more recent study(3) examined the dynamic homogeneity of brush macromolecules with different DP of the poly(n-butyl acrylate) side chains by dielectric spectroscopy and concluded that there is a certain degree of dynamic homogeneity induced by architecture. All short distance correlations in the bottle-brush molecules are dominated by the PBA side chains but long range correlations are dominated by the contrast between the backbone and brush side chains with an increasing separation between the backbones as the MW of the side chains increase with the maximum length corresponding to an all-trans conformation of the backbone being reached when the side chain MW is close to 12,500. When the DP of the side chains are only 30 or 50 the cylinder length per backbone monomer are lower and hence the backbone retains some conformational freedom.
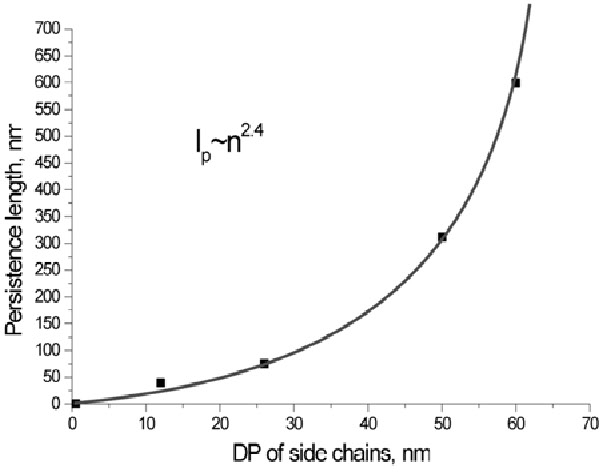
This is also the conclusion when the effect of the molecular weight of the side chain on the solution properties of bottle-brush macromolecules was measured under good solvent conditions with small-angle neutron scattering and static light scattering.(4) The systems under investigation were densely grafted brushes, synthesized via the “grafting-from” route, built from a poly(alkyl methacrylate) backbone to which poly(butyl acrylate) side chains were attached in the “grafting from” reaction. The aim of the work was to study how the systematic variation of structural parameters such as the side chain length and backbone length change the conformation of the polymer brushes in solution. All spectra can be consistently described by a model, considering the bottle-brush polymers as flexible rods with internal diameter fluctuations. The correlation between side chain length and persistence length of the backbone in molecular brushes is exponential. At longer side chain lengths (DP > 60), the persistence length approaches the length of the molecule; i.e. the backbone is fully extended.
In Langmuir Blodgett (LB) films, this translates to parallel organization over short length scales. A conformational collapse of the brush molecule can be induced by increasing the surface pressure on a Langmuir monolayer of the molecules on a water subphase.(5)

Transitions from extended to globular conformation and persistence lengths in extended conformation show strong dependence on grafting density. This transition was later examined in real time by monitoring the change in conformation by adsorption of ethanol or water from the contacting atmosphere.(6) This initial observation was examined in greater detail in later papers(7-8) where conformational changes were examined for brush macromolecules deposited on amorphous silicone oxide surfaces to avoid any interaction between the brush and the substrate. Exposure to alcohol vapor induced collapse of the individual molecules while exposure to water vapor promoted extension thereby supporting the original model of vapor/atmosphere induced conformational changes.
As the solution and bulk physical properties of these materials with complex intramolecular structure have begun to be examined a number of potential applications have presented themselves. The properties of the novel materials can be grouped by properties depending, to some degree, on the physical features of the molecules and properties tunable by composition. These include, solid stable replacements for hydrogels, (materials we have called “SuperSoft elastomers”) materials that provide templating environments for intramolecular chemistry such as directed mineralization of inorganic nanocrystals,(9) for the formation of high aspect ratio nanowires(10-12) and templates for carbon nanotubes(13) and stabilizers, surface-modifying agents, dispersants, emulsifiers, compatibilizers and impact resistant materials. They are model molecules for mechanochemistry and act as stimuli responsive materials (-pH, light and temperature). They also provide molecular fluidics and surface walking brushes.
SuperSoft Elastomers:
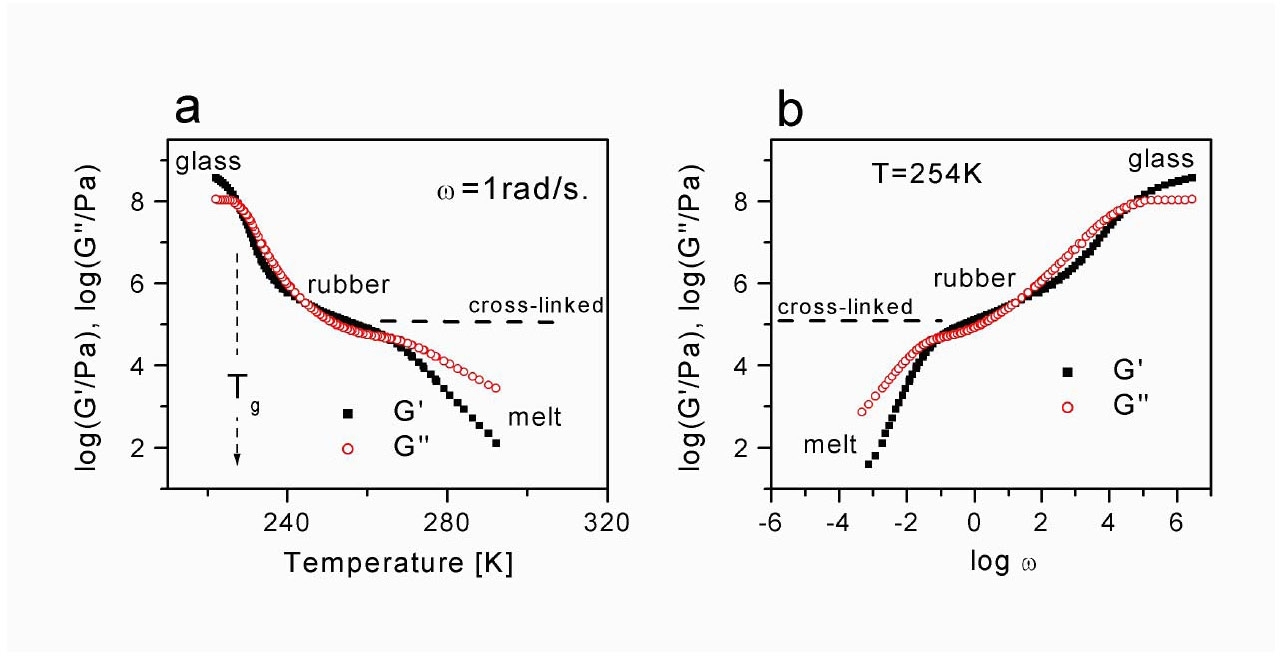
Common elastomeric materials have a typical Young's modulus value (at small strains) of the order of 105 to106 Pa, with reversible extensibility reaching 1000%. They are approximately five orders of magnitude softer, and three orders of magnitude more deformable than typical solids, Figs a and b, taken from Professor Pakula's talk, show results from dynamic mechanical spectroscopy for a typical thermoplastic polymer. The material transitions from a glass through segmental relaxation and chain relaxation to a flowing melt. Weakly cross-linked rubbers preserve the modulus of the rubbery plateau seen for the melt of linear entangled polymer chains (Mc remains similar to the order of Me), whereas, for highly cross-linked systems the modulus increases.
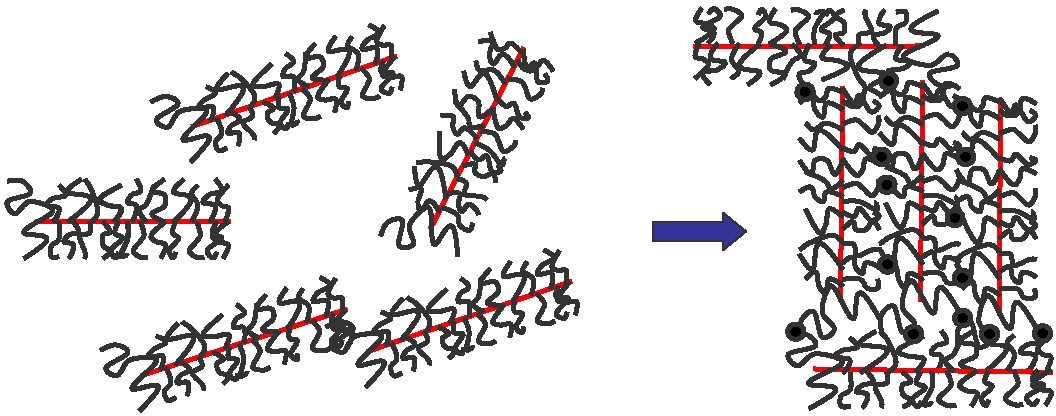
These unusual mechanical properties that had previously only been observed in aqueous gels are now observed for the first time in bulk polymeric materials. A possible phenomenological explanation is shown in the following schematic where • indicates the presence of an intramolecular chemical crosslink between two arms of different brush macromolecules forming a molecular network of crosslinked brush molecules which consists of linked backbone segments diluted by short tethered side chains.
The first materials recognized as precursors of super soft elastomers were bottle brush macromolecules with a very long backbone and densely grafted poly(n-butyl acrylate) side chains which displayed an ultra low modulus plateau in the soft gel range when dynamic mechanical spectroscopy was measured. When they were transformed to a network by chemical cross-linking, the material became a super-soft rubber(17) which instead of the expected elastomeric global flow range displayed a plateau in G’ extending towards low frequencies. This plateau indicates elastic properties for such polymers.
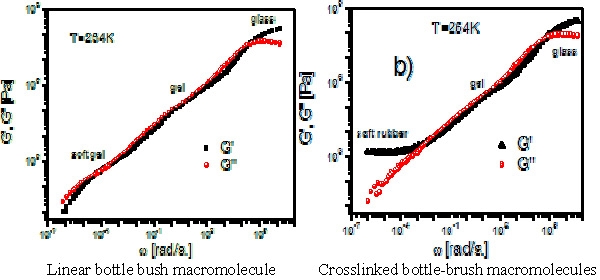
However in this case the plateau modulus is much lower than that seen for typical polymeric rubbers, which has to be attributed to the large fraction of the short dangling chains in the system acting as intrinsic plasticizers, making the material extremely soft.
The first bottlebrush copolymers that were converted into supersoft elastomers underwent the crosslinking reaction in the presence of residual copper present in the copolymer as a consequence of being prepared by ATRP. A more recent study set out to examine the effect of residual copper on the stability of molecular brushes prepared by atom transfer radical polymerization.(18) It was determined that although the copper concentration was decreased down to below ten ppm levels by passing through an alumina column, further removal by dialysis or precipitation was required to prevent crosslinking reactions.
Physical Crosslinking:
A SuperSoft thermoplastic elastomer can be formed when the backbone is selected to be an ABA block copolymer with phase separable A blocks and a bottle brush B segment. The properties of one such block copolymer are shown below.
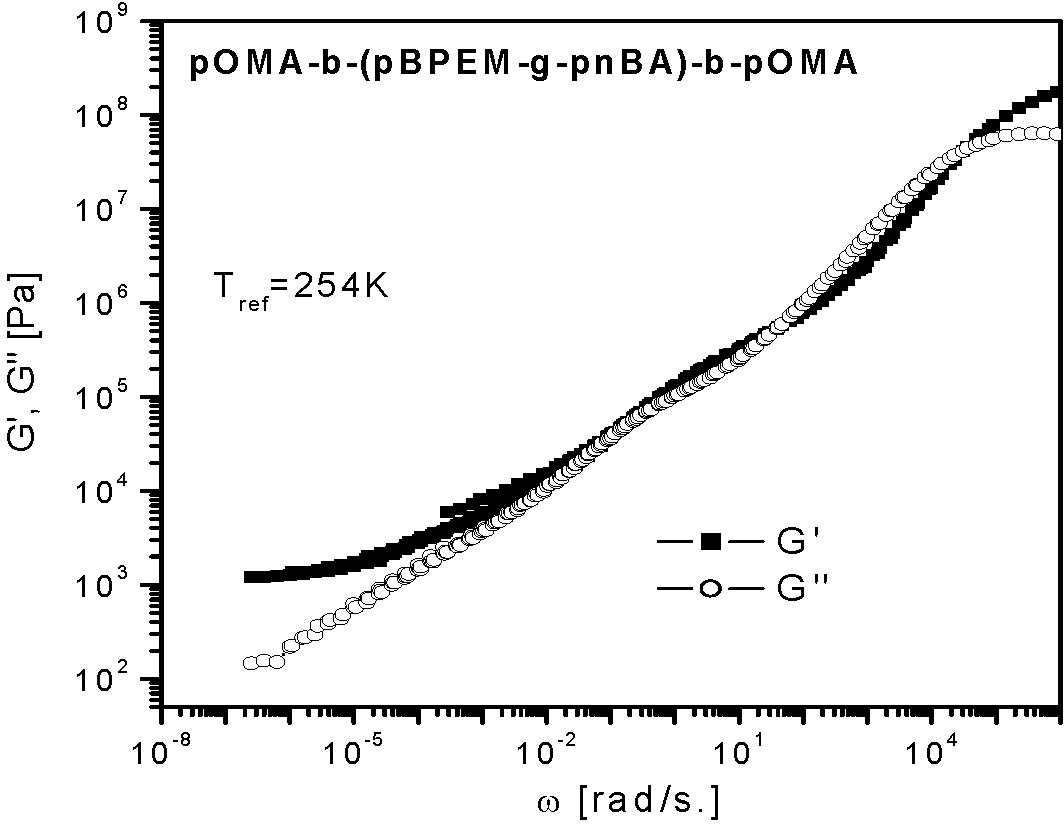
Frequency dependencies of the storage (G‘) and loss (G‘‘) modulus for the triblock copolymer with a brush-like middle block (master curves at the reference temperature of 254K). A discontinuity seen in G' corresponds to melting in the pOMA microphase taking place at 292K.
Therefore brush copolymers synthesized by controlled radical polymerization crosslinked either covalently or physically result in the synthesis of elastomers with an unusually low equilibrium shear modulus (G e) of the order 1 kPa. In a recent paper examples are given for both crosslink motifs, along with the dynamic viscoelastic properties of these materials.(19) The results are discussed in terms of the effect of the side chains on the brush polymers, which behave in some respect as low molecular weight diluents that cannot be leached from the sample.
The bulk tactile response can be modified by selecting the composition of the “dangling chains or hairs” and can vary from hydrophilic to hydrophobic and encompass attached oleophilic and oleophobic diluents that respond to environmental pressures. The bulk properties can be further modified, as discussed above, by changing the degree of cross-linking of the backbone network to form a material with the desired modulus for the targeted application. Higher levels of cross-linking, or increased stiffness of either the backbone or tethered dangling graft or “hair” will increase the modulus while still providing polymers significantly softer than current elastomeric materials.
Bond Scission:
Covalent carbon-carbon bonds are hard to break. Their strength is evident in the hardness of diamonds and tensile strength of oriented polymeric fibers. On the single-molecule level, it manifests itself in the need for forces of several nano-newtons to extend and mechanically rupture one bond. Such forces have been generated using extensional flow, ultrasonic irradiation, receding meniscus and by directly stretching a single molecule with nano-probes.
It was shown that simple adsorption of brush-like macromolecules with long side chains on a substrate can induce not only conformational deformations, but also spontaneous rupture of covalent bonds in the macromolecular backbone.(18) This behavior can be attributed to the fact that the attractive interaction between the side chains and the substrate is maximized by the spreading of the side chains, which in turn induces tension along the polymer backbone. Provided the side-chain densities and substrate interaction are sufficiently high, the tension generated will be strong enough to rupture covalent carbon-carbon bonds. A molecule where each side chain has 140 monomeric units and an energy change upon adsorption: Eadsorption~n´kT = 140 kT which is comparable to the energy of a C-C bond: EC-C~150 kT led to chain scission. The following images show how a brush molecule with a DP of the backbone = 400 and DP of each side chain = 140 undergoes progressive chain scission when the polymer is dispersed on the surface of a water/propanol (99.8/0.2 wt/wt%) solution.

The rate constant for C-C bond cleavage was shown to be extremely sensitive to the substrate surface energy.(21) A few percent increase in the surface energy from 69.2 to 71.2 mN/m led to an order of magnitude increase of the scission rate. The absolute values of the forces required to rupture the C-C bond ranging from 2.57 to 2.47 nN are in agreement with previously calculated and measured values for stretching surface-tethered molecules. However unlike conventional chemical reactions, the rate of bond scission was shown to decrease with temperature.(22) This apparent anti-Arrhenius behavior was caused by a decrease in the surface energy of the underlying substrate upon heating, which results in a corresponding decrease of bond tension in the adsorbed macromolecules. Even though the tension dropped minimally from 2.16 to 1.89 nN, this was sufficient to overpower the increase in the thermal energy (kBT) in the Arrhenius equation.
We expect similar adsorption-induced backbone scission to occur for all macromolecules with highly branched architectures, such as dendrimers. This behavior needs to be considered when designing surface-targeted macromolecules of this type either to avoid undesired degradation, or to ensure rupture at predetermined macromolecular sites. Indeed we designed brush-like macromolecules that are able to focus tension to a single disulfide bond within the polymer backbone leading to its scission while the other bonds remain intact.(23) Bond tension is spontaneously generated within brushlike macromols. as they spread on a solid substrate. The molecular architecture creates an uneven distribution of tension in the covalent bonds, leading to spatially controlled bond scission. By controlling the flow rate and the gradient of the film pressure, one can sever the flowing macromolecules with high precision. Specific chemical bonds are activated within distinct macromolecules located in a defined area of a thin film. Furthermore, the flow-controlled loading rate enables quantitative analysis of the bond activation parameters.
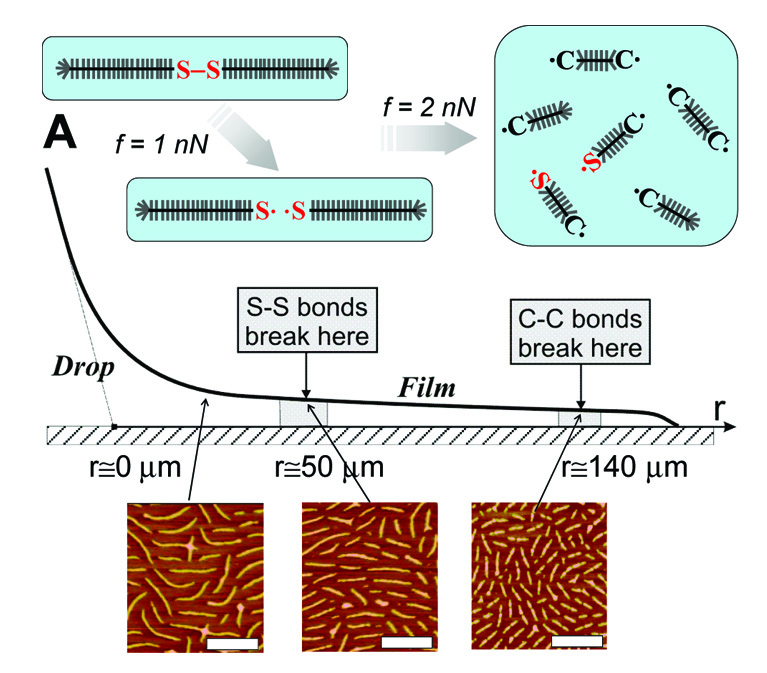
The above schematics from reference (24) illustrates the degradation process of molecular brushes having a disulfide bond in the middle of the backbone as the film spreads from a drop of a polymer melt is deposited on a mica substrate and is allowed to flow in a controlled environment. During flow, the initial rupture of the weaker disulfide linker is followed by random scission of carbon-carbon bonds. The height AFM images are captured at three different distances from the drop edge (r = 20, 50, and 140 μm) to demonstrate the mid-chain S-S fracture of the brushlike macromolecules followed by the scission of the C-C bonds. This corroborates that the degradation process in the low-tension areas (ca. 1 nN) is dominated by the scission of the disulfide linker (ES-S ) 268 kJ/mol) proceeding at a much faster rate compared to the stronger C-C bonds (EC-C) 347 kJ/mol) in the same backbone. The onset of scission of the C-C bonds occurs at larger distances from the drop at 140 μm, where the bond tension reaches a magnitude of 2 nN. The spreading-induced tension in the polymer backbone probes the strength of individual bonds triggering scission of the weaker ones. This indicates that these molecules can be used as a tool for mechanical activation of chemical reactions at specific chemical bonds.(23)
Bottlebrush macromolecules containing a disulfide linker in the middle of the backbone were also employed as miniature tensile machines to study the effect of mechanical force on the kinetics of disulfide reduction by dithiothreitol (DTT). The scission reaction was monitored through molecular imaging by AFM and it was determined that the scission rate constant increases linearly with the concentration of DTT and exponentially with mechanical tension exerted on the disulfide bond. Moreover, the rate constant at zero force was found to be significantly lower than the reduction rate constant in bulk solution which suggests an acidic compound on the water surface with pH = 3.7. This work demonstrates the ability of branched macromolecules to accelerate chemical reactions at specific covalent bonds without applying an external force.
Some Other Properties of "Bottle Brush" Molecules:
The properties of individual brush macromolecules are beginning to be examined in detail. A new type of flow fingering instability was observed in monolayer-thick polymer films as they spread on a solid substrate.(25) Tracing the movement of individual molecules by AFM allowed observation of individual molecules and one could follow the development of the flow instability on the molecular level. This led to an understanding of the underlying physical mechanism. The fingering instability was observed to be triggered by conformational changes of the brush-like macromolecules in response to the pressure gradient driving the flow as a drop of solution gradually spread out on the surface.
Spreading of homogeneous mixtures of bottle-brush and linear macromolecules of poly(n-butylacrylate) on a solid substrate was monitored on the molecular scale by AFM.(26) Despite the nearly identical chemical composition and similar MW the brush-like macromolecules move markedly slower than linear chains. Moreover, smaller bottle-brushes were shown to flow faster than the larger bottle-brushes, resulting in a size fractionation of the macromolecules along the spreading direction. This behavior was explained by the difference in sliding friction coefficient between the bottle-brush macromolecules and linear chains with the substrate. A theoretical model of molecular size separation is in a good agreement with the experimental data.
On another page we indicated that brush block copolymers where the backbone of the brush copolymer was a block copolymer could be prepared by a combination of several controlled polymerization procedures.(27) When the self-assembly of PS-PLA bottlebrush block copolymers with varying lengths of branches and backbones were examined a number of unusual trends, which were attributed to the dynamic, three-dimensional structure of the brush block copolymer, were detected. The materials formed lamellae microstructures which results suggested that in the phase-separated melts the bottlebrush block copolymer backbone, while still in a chain extended conformation, still possesses a certain degree of flexibility necessary to accommodate different interfacial areas and pack into lamellae microstructures.
Ionic Conductors:
Grafted PEO brush polymers were examined as potential solvent-free, lithium ion conducting materials. The copolymers were doped with CF3SO3-Li+.
Analysis of the structure, mechanical properties and ionic conductivity of these systems in bulk using mechanical spectroscopy and dielectric spectroscopy showed that the specific architecture and the presence of lithium ions suppresses completely the crystallization of PEO side chains graft segments. Consequently, amorphous homogeneous materials are obtained in which both high local mobility and sufficient macroscopic mechanical stability are achieved at the same time. When melts of linear brush polymers were optimally doped with CF3SO3–Li+ ionic conductivity reached 10-3 S/cm at temperatures close to room temperature.(28)
Cadmium selenide (CdSe) nanowires were successfully fabricated in situ by utilizing amphiphilic core-shell cylindrical polymer brushes as well-defined single molecule templates.(11) The hydrophilic polymer brush core acts as a nano-reactor for generating and shaping complexed CdSe nanoparticles into nanowires via the absorption of cadmium ions by carboxylate groups in the core and subsequent introduction of H2Se gas. The hydrophobic polymer brush shell protects the nanowires from agglomeration and renders the hybrid a soluble material. The formation of 170 nm-long CdSe nanowires proceeds simultaneously with the nucleation, growth and combination of CdSe nanoparticles. After the introduction of CdSe nanowires, the recuperated chemical structure of core-shell cylindrical polymer brushes facilitates a double-loading process, broadening the CdSe nanowire from 7.5 to 9.3 nm on an average Both hybrids are soluble and stable in organic solvents for one year and and therefore are potential candidates for preparation of nanoscale optic and electronic devices.
Phototunable:
Molecular brushes with trans-4-methacryloyloxyazobenzene (MOAB) and 2-(dimethylamino)ethyl methacrylate (DMAEMA) units in the side chains were successfully synthesized by grafting from a poly(2-(2-bromopropionyloxy)ethyl methacrylate) (PBPEM) macroinitiator. Molecular weight, molecular weight distribution, and the DP of the side chains containing different distributions of DMAEMA and MOAB were detected by gel permeation chromatography (GPC) and 1H NMR spectroscopy.
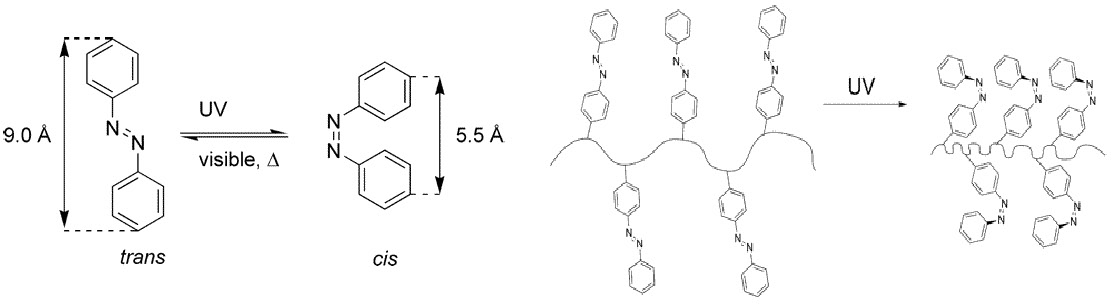
The number-average molecular weight (Mn) of the brushes ranged from 4.7 X 105 to 1.1 X 106 depending on molecular architecture. The molecular weight distribution of the brushes was narrow (Mw/Mn = 1.23-1.36). UV-vis spectra of the brush copolymers in either chloroform or aqueous solution showed reversible isomerization of azobenzene units in the side chains upon irradiation with UV (365 nm) or visible light (442 nm).(29)
The transmission spectra of the aqueous solutions of the brush copolymers at 600 nm were measured as a function of temperature and showed that the lower critical solution temp. (LCST) can be affected by photo-irradiation.
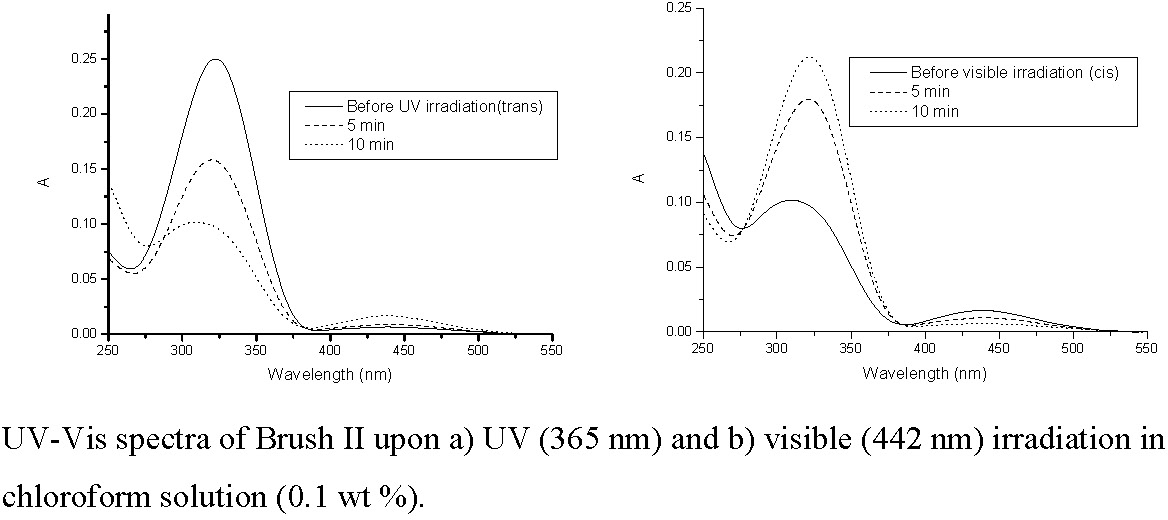
Upon irradiation with UV light, the azobenzene groups of the MOAB units undergo a trans to cis isomerization that is accompanied by an increase in hydrophilicity. The increased solubility of the MOAB units in response to photo-stimulation resulted in brushes with temperature and light-responsive behavior. The following figure shows % transmission at 600 nm of an aqueous solutions of poly[BPEM-g-(DMAEMA-stat-MOAB)] as a function of temperature. The LCST (indicated by decrease in transmission) is dependent on the isomerization of the azobenzene groups in the MOAB units. The particle size in aqueous solution measured by dynamic light scattering ranged from 30 to 120 nm depending on temperature, concentration, and structural state of the azobenzene units.
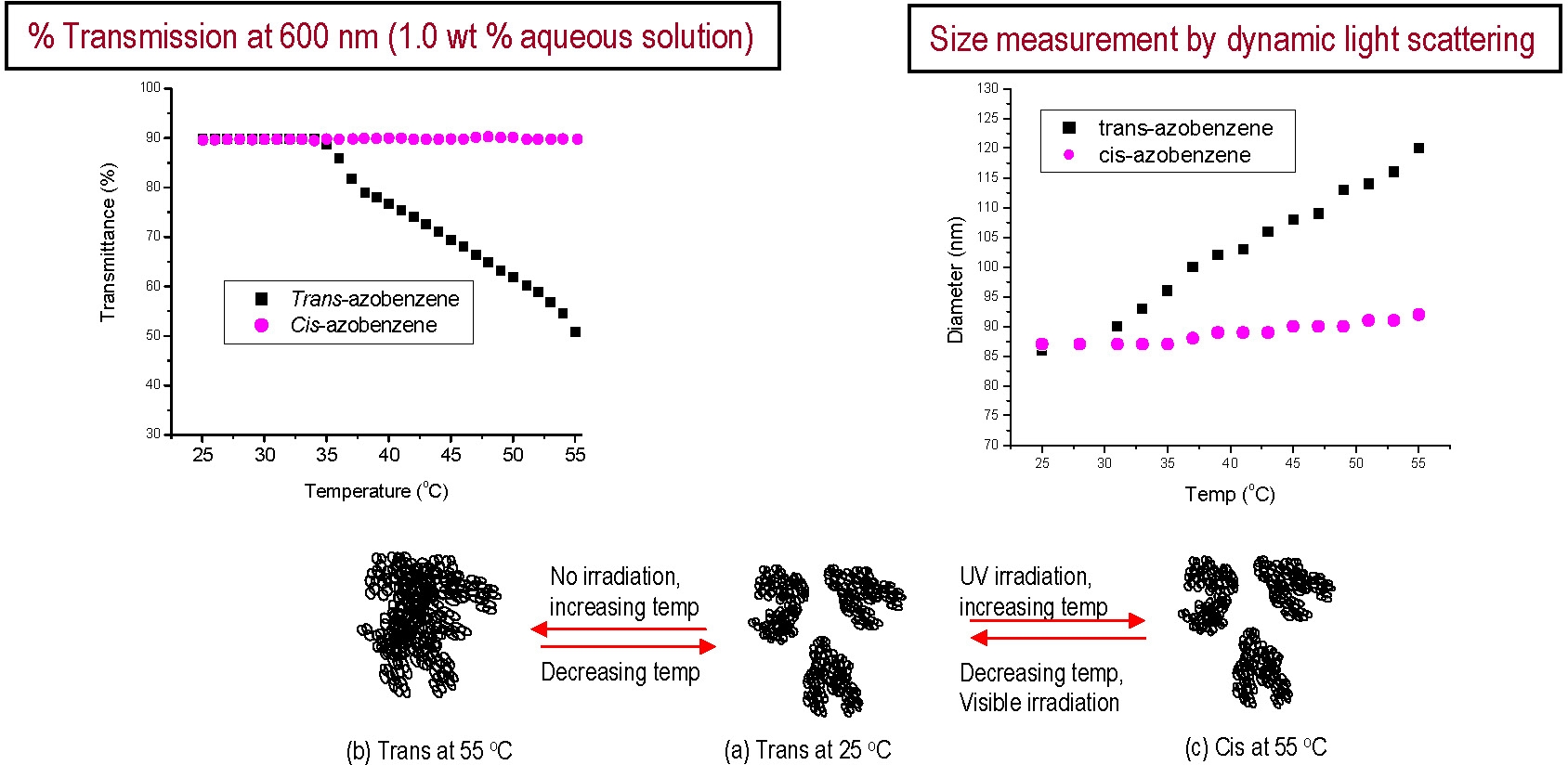
This variation of particle size gave additional evidence for photoresponsive thermal transition.
Crystallizable:
Poly(ethylene glycol) methy ether methacrylate macromonomer (PEOMA, MW = 1100 g/mol, DP PEO = 23) and octadecyl methacrylate or acrylate (ODMA, ODA) were (co)polymerized by ATRP. The one-pot copolymerization via grafting through (macromonomer method) yielded densely heterografted copolymers (DP (n) = 300-500).(30) The composition of the copolymers strongly depended on the selection of comonomer pairs; i.e., two methacrylates (PEOMA/ODMA) or methacrylate/acrylate (PEOMA/ODA) led to formation of a spontaneous random or gradient copolymer, respectively. The combination of hydrophilic and hydrophobic segments in the same polymer chain caused microphase segregation. Additionally, x-ray results indicated that frustration in packing the crystallizable segments, helical PEOMA and hexagonal ODMA crystals, can direct to an amorphous fraction instead of a semicrystaline ODMA in the graft copolymer. Thermomechanical measurements showed the soft rubbery behavior of the copolymers (104 > G' > 103 Pa, G' > G''). The synthesis of homopolymers with a high degree of polymerization is also reported (DP PPEOMA > 325, DP PODMA > 435, DP PODA > 150).
A molecular brush with block copolymer side chains, i.e., a crystalline poly(ε-caprolactone) (PCL) core with an amorphous poly(n-butyl acrylate) (PBA) shell, was synthesized by the “grafting from” approach using a combination of ring-opening polymerization (ROP) and atom transfer radical polymerization (ATRP).(31) In the following AFM height and phase images the macromolecules have clearly delineated and extended backbones resulting from side-chain desorption and repulsion providing proof of chain extension. More importantly, one observes the presence of distinct stripes within the corona of the macromolecule. The stripes are attributed to a very peculiar microphase separation of the crystalline PCL core blocks and amorphous PBA corona blocks. This system is very frustrated since the phase separation is confined within one molecule, and it is strongly constrained due to the covalent connectivity of the side chains. These constraints may result in adsorption of PBA blocks on the surface of the adsorbed PCL core blocks or crystallization of PCL blocks on top of the layer of PBA segments.

Subsequently an understanding of the crystallization phenomenon of molecular brushes with block copolymer side chains individually, in a film as well as in bulk was obtained.(32) The molecular packing model revealed the unique crystallization behavior of molecular brushes with block copolymer side chains in a thin film or bulk. A series of three cylindrical molecular brushes with poly(ε-caprolactone)-b-poly(butyl acrylate) side chains were investigated with regard to the effect of the intramolecular confinement on the crystalization behavior of the PCL core block. While the length of the PCL block was maintained (nPCL = 50), the DP of the PBA corona block was varied from nPBA = 0 (PCL brush) to nPBA = 52 and 163. The crystalization behavior of the studied polymers was shown to be different in thin films and bulk samples.
In thin films, the brushlike macromols, were fully extended and remained segregated due to the steric repulsion of the adsorbed PBA corona. Under the constraint of the backbone extension, crystalization of of the PCL core led to formation of a characteristic spinelike morphology.


Single brush molecules of PCL (A) and brush with block copolymer side chain with DP of PBA -150 (B) spincast on mica. The homopolymer brushes (A) exhibit the conventional wormlike morphology, while the brushes with block copolymer side chains (B) reveal a distinctive spinelike morphology with short ribs evenly distributed along the backbone. The larger scale images of thicker films of the homopolymer brush (C) and side chain block copolymer (D) samples, prepared by the Langmuir-Blodgett technique, reveal that the block copolymer sample has a significant fraction of longer molecules compared to the homopolymer sample. The corresponding length distributions are consistent with this observation, which is attributed to the end-to-end association of PCL-b-PBA brushes.
In bulk samples, the core-shell confinement did not prohibit breakout crystalization leading to the formation of a spherulitic morphology similar to that observed for linear and brushlike PCL. However, the crystalization process of the PCL-b-PBA brushes was significantly slower as compared to the linear and pure PCL-brushlike counterparts as it evolved through a transition from the molecularly segregated core-shell morphology to a lamellar organization of multiple molecules.
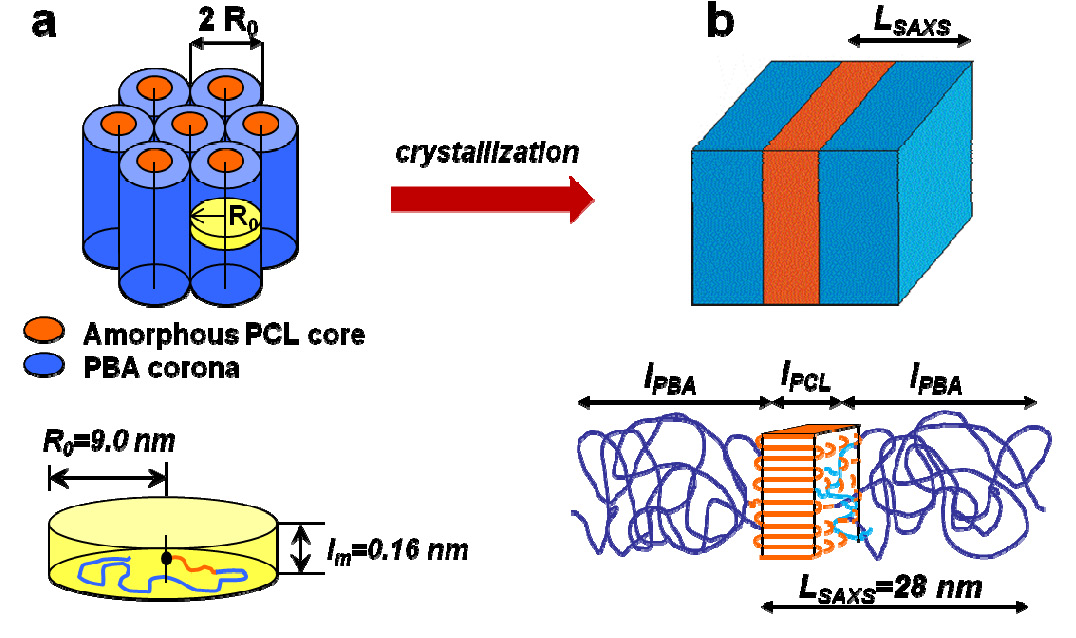
The above schematic shows the molecular packing in thick films and bulk (A) before and (B) after crystallization of the PCL core. The indicated dimensions correspond to sample the sample with PBA segments of DP 150. In the amorphous state, the molecules can be modeled as microphase-separated cylinders with a PCL core and PBA shell with a diameter of 2R0. Upon crystallization, the initially cylindrical morphology is disrupted by the crystallization of PCL. The block copolymer side chains can be modeled as rectangular lamellae with a thickness LSAXS - the long period from SAXS. The rectangle is composed of two layers: folded chain PCL lamellae and an amorphous layer which includes both the PBA blocks (dark blue) and the PCL disordered segments (light blue). The brush backbone (light blue) is assumed to segregate at the interface between the two layers to decrease the associated perturbation of the crystalline structure. Crystallization of the PCL block is evidenced by the development of the second peak at LSAXS = 28 nm (Fig. 6D). As shown in Scheme 2b, this spacing is attributed to the long period of alternating crystalline and amorphous layers
Drawing of fibers containing nascent crystallites resulted in the final morphology composed of crystaline lamellae parallel to the fiber axis with the PCL blocks oriented perpendicular to the axis. The lamellae thickness was shown to decrease with the length of the amorphous PBA block.
Stimuli Responsive Molecules:
The first stimuli responsive water soluble molecular brush was prepared by Manfred Schmidt(33) who prepared brushes with poly(N-isopropylacrylamide) (PNIPAM) side chains. This is one of the few published examples of the preparation of water-soluble brushes via a "grafting from" process. The polymers underwent a thermally induced collapse from extended cylindrical structure to spheres. We prepared temperature-responsive brushes with tunable LCST's by statistical copolymerization of DMA and n-butyl acrylate from a macroinitiator backbone. A similar behavior, collapse of the extended cylinder to a sphere was envisioned.
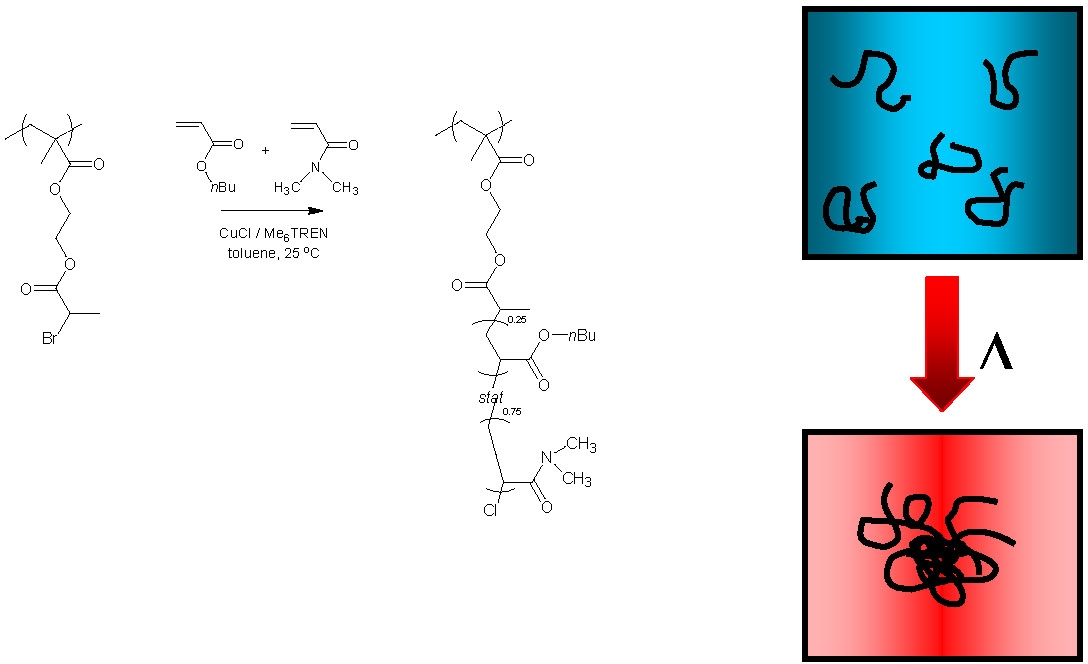
However when a 1.0 w% solution of the brush was warmed through the LCST an increase in volume was observed as a function of temperature, with DH = 57 nm at 20 °C leading to DH = 104 nm at 55 °C. The broad nature of the transition is common for statistical copolymers that demonstrate LCST behavior and is most likely a result of composition polydispersity. The increase in size for species in solution is typical of an LCST transition and is the result of intermolecular aggregation. Interestingly, when the concentration of the copolymer in the solvent was decreased by an order of magnitude, to 0.1 w%, the opposite behavior was observed in that the hydrodynamic diameter decreased as a function of temperature (DH = 47 nm at 55 °C).
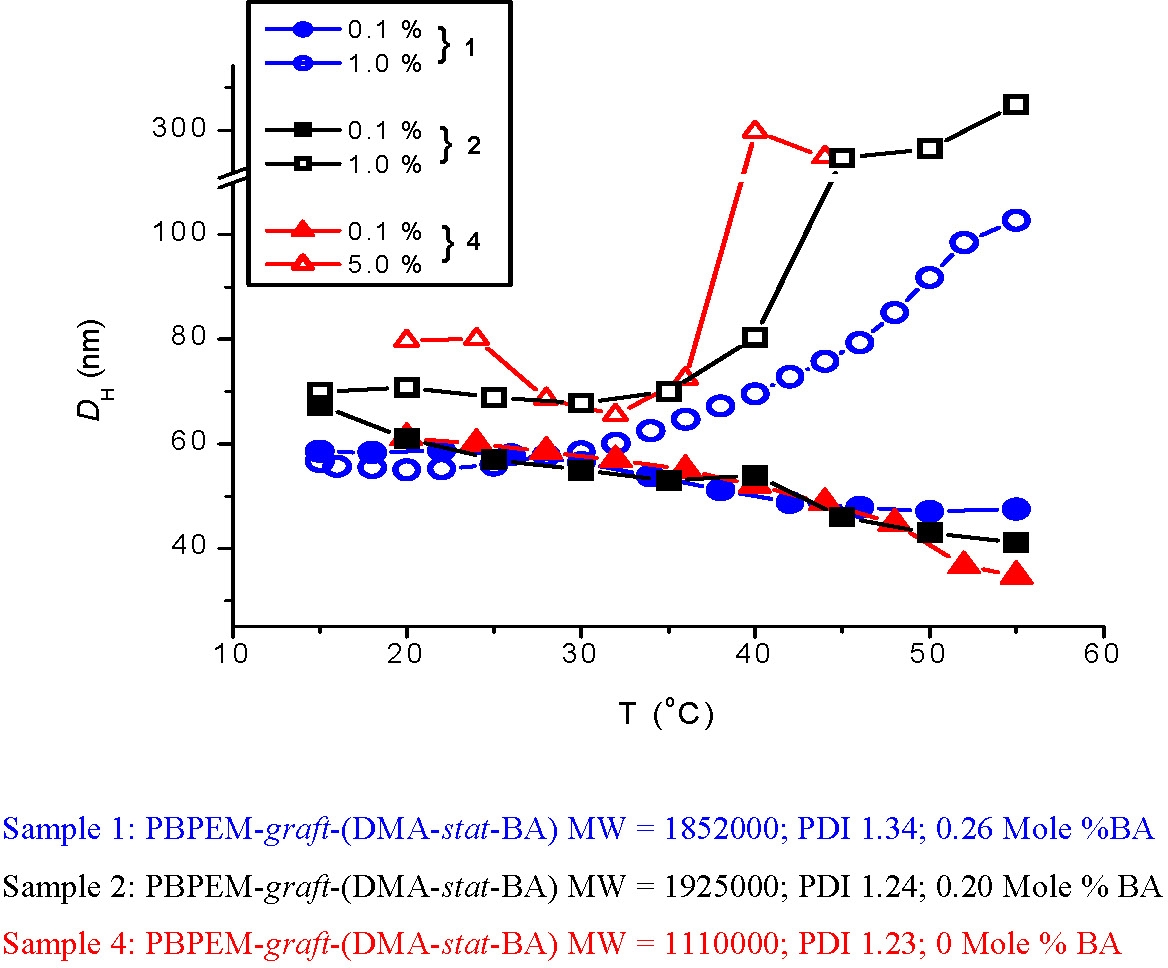
This was attributable to higher solution concentrations favoring dehydration and subsequent intermolecular aggregation, typical for (co)polymers that demonstrate LCST behavior but, when the solution is diluted, side chains dehydrate on heating, and intramolecular collapse is favored, such that individual brushes transition from extended cylinders to compact spheres.(34)
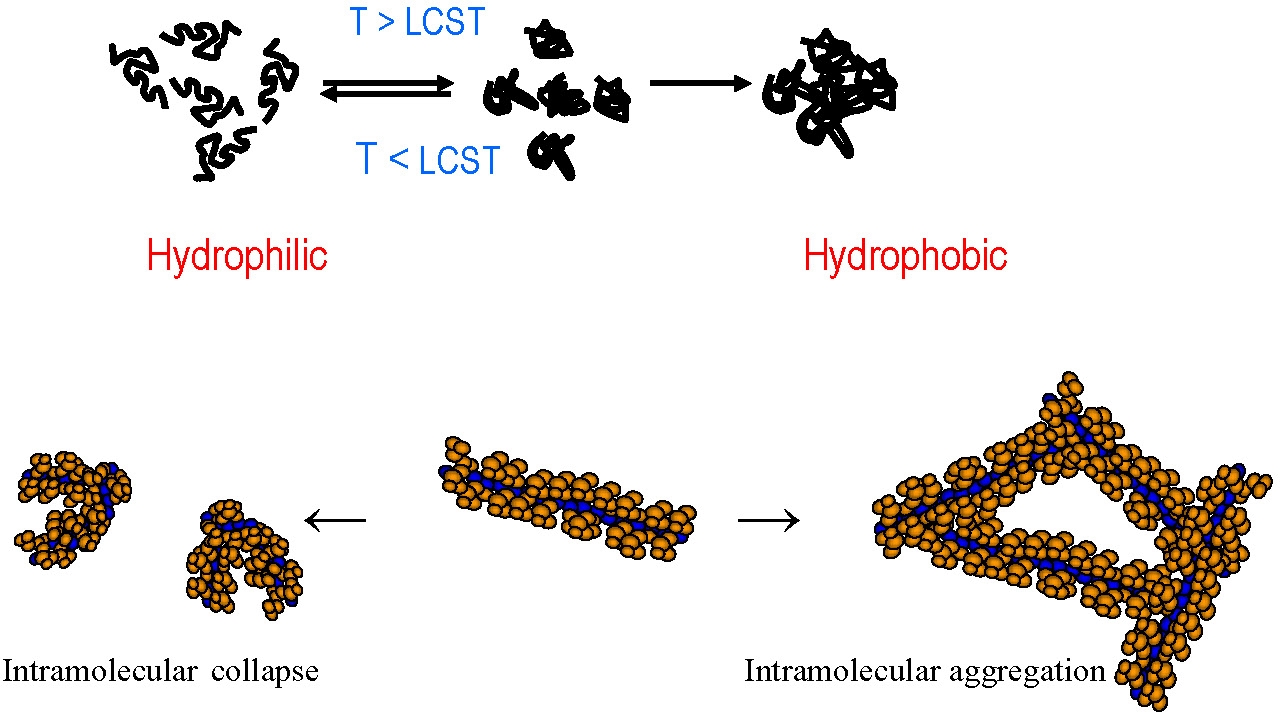
Thermoresponsive brushes were also prepared with side chains consisting of statistical copolymers of di(ethylene glycol) Me ether methacrylate (MEO2MA) and tri(ethylene glycol) Me ether methacrylate (MEO3MA).(35) The lower critical solution temperature (LCST) of the brushes increased with the mole fraction of MEO3MA in the side chain, and the hysteresis between the heating and cooling cycles decreased with the length of the side chain. Brush copolymers with different graft density were also prepared. The average hydrodynamic diameter, measured by dynamic light scattering (DLS), varied with temperature above the LCST, and the maximum diameter of the aggregates increased according to the graft density of side chain along the brush backbone. These two monomers were also incorporated into side chain block copolymer brushes by ATRP. The cloud point of solutions of the block brushes displayed two stages of aggregation during heating, exhibiting the results of both intermolecular and intramolecular aggregation. This behavior was strongly dependent on the sequence of the side chain segments. As the temperature increased, particles consisting of collapsed PMEO2MA and PMEO3MA segments aggregated upon further heating to precipitate as larger particles.
pH Responsive Materials:
A series of loosely grafted water-soluble poly(acrylic acid) (PAA) brushes with four different grafting densities were synthesized by the "grafting from" approach. AFM was used to study the conformation of adsorbed brushes as a function of pH. The adsorbed molecules undergo a globule-to-extended conformational transition as the pH of the solution is changed from acidic to basic. This transition was monitored on a mica surface by imaging individual molecules and the conformational behavior was compared with 100%-grafted PAA brushes. Unlike the loose brushes, the 100%-grafted molecules remained fully extended over a broad range of pH values (pH = 2-9) due to steric repulsion between the densely grafted side chains which is strongly enhanced upon adsorption to a substrate.(36)
pH-responsive fluorescent molecular bottlebrushes were prepared by the copolymerization of fluorescein O-methacrylate with n-butyl acrylate in a "grafting from" reaction.(37) Fluorescein is nontoxic, highly soluble in water in addition to being pH responsive and it was thought that the final product should be good for biomedical applications.
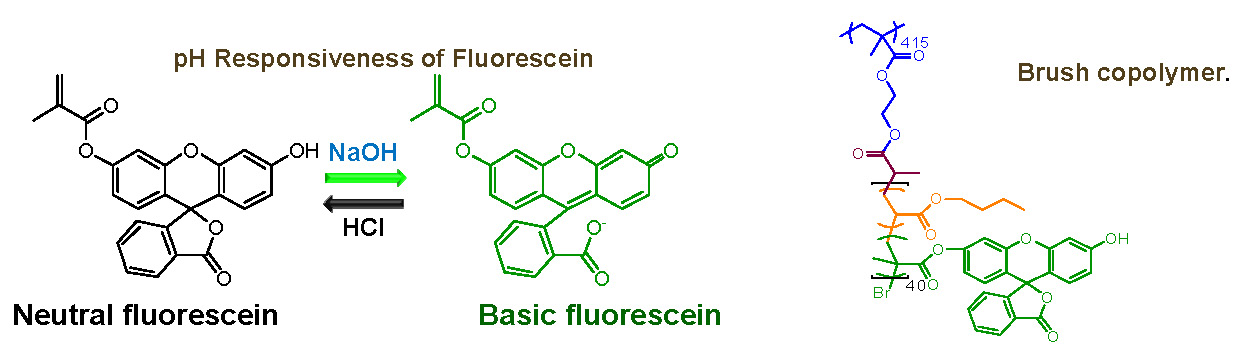
The structure of the synthesized brush macromolecules was confirmed through molecular imaging by AFM and NMR spectroscopy showed that the fluorescent units were successfully incorporated into the polymer. The synthesized bottlebrushes displayed highly fluorescent properties under basic conditions, yet showed no fluorescence under neutral or acidic conditions. The fluorescence could be turned on and off by changing the pH of the solution.
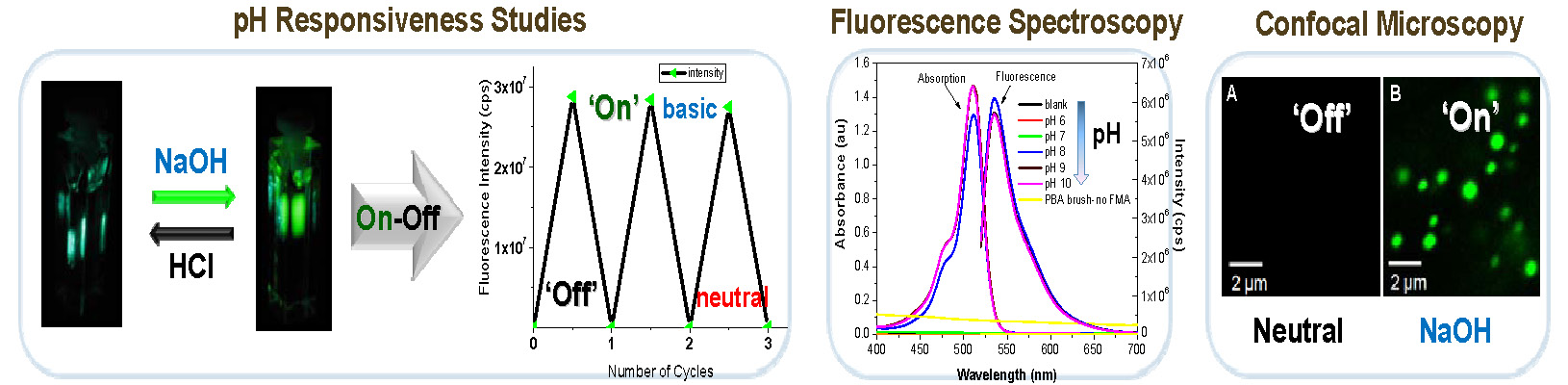
Dual Responsive Materials:
Brushes consisting of statistical copolymers of di(ethylene glycol) Me ether methacrylate (MEO2MA) and either methacrylic acid (MAA) or N,N-dimethylaminoethyl methacrylate (DMAEMA) were synthesized by grafting from poly(2-(2-bromoisobutyroyloxy)ethyl methacrylate) (PBIEM) macroinitiators using ATRP.(35, 38) The lower critical solution temperature (LCST) of MEO2MA-stat-MAA decreased with the increasing molar fraction of MAA in the copolymer in deionized water but increased in buffer solution at pH 7. At pH 9, the hydrophilicity of polymer increased with ionization of carboxylic acid to further raise the LCST. The LCST of MEO2MA-stat-DMAEMA copolymers increased with increasing DMAEMA content at pH 4, 7, and 9. A bottle-brush terpolymer prepared from all three comonomers exhibited LCST at pH 4 and 7 but not at pH 9, which can be attributed to the stronger ionization of MAA. The responsive nature of the copolymer is enhanced by the densely graft structure of a brush copolymer.
Repeatable Fibrillar Adhesives
Repeatable adhesive materials were prepared by controlled grafting of dangling poly(butyl acrylate) (PBA) chains from polymer elastomers. The dangling chainelastomer system was prepared by grafting butyl acrylate from a prefunctionalized polydimethylsiloxane (PDMS) elastomeric network.(39) To study the effects of chain growth and network strain as they relate to network adhesion mechanics, various lengths of PBA chains with degree of polymns. (DP) of 65, 281, 508, and 1200 were incorporated into the PDMS matrix. PBA chains with a DP value of 281 grafted from a flat PDMS substrate showed the highest, approximately 3.5-fold, enhancement of nano-and macroscale adhesion relative to a flat raw, ungrafted and not prefunctionalized, PDMS substrate. Moreover, to study the effect of PBA dangling chains on adhesion in fibrillar elastomer structures inspired by gecko foot hairs, a dip-transfer fabrication method was used to graft PBA chains with a DP value of 296 from the tip endings of mushroom-shaped PDMS micropillars. The PBA chain covered micropillar array showed macroscale adhesion enhancement up to approximately 7 times relative to the flat ungrafted pre-functionalized PDMS control substrate, showing additional non-optimized approximately 2-fold adhesion enhancement due to fibrillar structuring and mushroom shaped tip ending. These dangling hetero chains on elastomer micro-/nanofibrillar structures may provide a novel fabrication platform for multilength scale, repeatable, and high-strength fibrillar adhesives inspired by gecko foot hairs.A recent collaborative paper provides a review of the preparation and properties of stimuli-responsive molecular brushes.(40) Brush and brush block macromolecules that displayed a change in conformation as well as changes in physical properties of polymers by external stimuli, such as solvent, ionic environment, temperature, pH, mechanical stress and light were discussed in addition to materials exhibiting a combination of incorporated external stimuli response functions. The driving force for this research is the increasing need for “smartness” in biomedical and engineering materials which has generated a growing interest for synthetic polymers that exhibit environmentally responsive behavior.
REFERENCES
(1) Sheiko, S. S.; Sumerlin, B. S.; Matyjaszewski, K. Prog. Polym. Sci. 2008, 33, 759-785.
(2) Pietrasik, J.; Sumerlin, B. S.; Lee, H.-i.; Gil, R. R.; Matyjaszewski, K. Polymer 2007, 48, 496-501.
(3) Grigoriadis, C.; Nese, A.; Matyjaszewski, K.; Pakula, T.; Butt, H.-J.; Floudas, G. Macromol. Chem. Phys. 2012, 213, 1311–1320,.
(4) deGennes, P. G.; Cornell University: Ithaca, 1979.
(5) Sheiko, S. S.; Prokhorova, S. A.; Beers, K. L.; Matyjaszewski, K.; Potemkin, I. I.; Khokhlov, A. R.; Moeller, M. Macromolecules 2001, 34, 8354-8360.
(6) Gallyamov, M. O.; Tartsch, B.; Khokhlov, A. R.; Sheiko, S. S.; Boerner, H. G.; Matyjaszewski, K.; Moeller, M. Chemistry--A European Journal 2004, 10, 4599-4605.
(7) Gallyamov, M. O.; Tartsch, B.; Mela, P.; Boerner, H.; Matyjaszewski, K.; Sheiko, S.; Khokhlov, A.; Moeller, M. Physical Chemistry Chemical Physics 2007, 9, 346-352.
(8) Gallyamov, M. O.; Tartsch, B.; Mela, P.; Potemkin, I. I.; Sheiko, S. S.; Borner, H.; Matyjaszewski, K.; Khokhlov, A. R.; Moller, M. Journal of Polymer Science, Part B: Polymer Physics 2007, 45, 2368-2379.
(9) Kiriy, A.; Gorodyska, G.; Minko, S.; Jaeger, A. S., P.; Stamm, M. J. Amer. Chem Soc. 2002, 124, 13454-13462.
(10) Djalali, R.; Li, S.-Y.; Schmidt, M. Macromolecules 2002, 35, 4282-4288.
(11) Yuan, J.; Drechsler, M.; Xu, Y.; Zhang, M.; Mueller, A. H. E. Polymer 2008, 49, 1547-1554.
(12) Yuan, J.; Xu, Y.; Walther, A.; Bolisetty, S.; Schumacher, M.; Schmalz, H.; Ballauff, M.; Mueller, A. H. E. Nature Materials 2008, 7, 718-722.
(13) Tang, C.; Dufour, B.; Kowalewski, T.; Matyjaszewski, K. Macromolecules 2007, 40, 6199-6205.
(14) Rathgeber, S.; Pakula, T.; Wilk, A.; Matyjaszewski, K.; Beers, K. L. Journal of Chemical Physics 2005, 122, 124904/124901-124904/124913.
(15) Inoue, T.; Yamashita, Y.; Osaki, K. Macomolecules 2002, 35, 1770.
(16) U.P.Schröder; Oppermann, W. Physical properties of polymer gels; John Wiley & Sons: Chichester, 1996.
(17) Pakula, T.; Matyjaszewski, K. In PCT Int. Appl.; (Carnegie Mellon University, USA). WO 2004014963, 2004; p 65 pp.
(18) Nese, A.; Sheiko, S. S.; Matyjaszewski, K. Eur. Polym. J. 2011, 47, 1198-1202.
(19) Pakula, T.; Zhang, Y.; Matyjaszewski, K.; Lee, H.-i.; Boerner, H.; Qin, S.; Berry, G. C. Polymer 2006, 47, 7198-7206.
(20) Sheiko, S. S.; Sun, F. C.; Randall, A.; Shirvanyants, D.; Rubinstein, M.; Lee, H.-i.; Matyjaszewski, K. Nature 2006, 440, 191-194.
(21) Lebedeva, N. V.; Sun, F. C.; Lee, H.-i.; Matyjaszewski, K.; Sheiko, S. S. J. Am. Chem. Soc. 2008, 130, 4228-4229.
(22) Lebedeva, N. V.; Nese, A.; Sun, F. C.; Matyjaszewski, K.; Sheiko, S. S. Proc. Natl. Acad. Sci. U. S. A. 2012, 1-5.
(23) Li, Y.-C.; Nese, A.; Lebedeva, N. V.; Davis, T.; Matyjaszewski, K.; Sheiko, S. S. J. Am. Chem. Soc. 2011, 133, 17479–17484.
(24) Park, I.; Shirvanyants, D.; Nese, A.; Matyjaszewski, K.; Rubinstein, M.; Sheiko, S. S. J. Am. Chem. Soc. 2010, 132, 12487-12491.
(25) Xu, H.; Shirvanyants, D.; Beers, K. L.; Matyjaszewski, K.; Dobrynin, A. V.; Rubinstein, M.; Sheiko, S. S. Physical Review Letters 2005, 94, 237801/237801-237801/237804.
(26) Barrett, M. J.; Sun, F. C.; Nese, A.; Matyjaszewski, K.; Carrillo, J.-M. Y.; Dobrynin, A. V.; Sheiko, S. S. Langmuir 2010, 26, 15339-15344.
(27) Rzayev, J. Macromolecules 2009, 42, 2135-2141.
(28) Zhang, Y.; Costantini, N.; Mierzwa, M.; Pakula, T.; Neugebauer, D.; Matyjaszewski, K. Polymer 2004, 45, 6333-6339.
(29) Lee, H.-i.; Pietrasik, J.; Matyjaszewski, K. Macromolecules 2006, 39, 3914-3920.
(30) Neugebauer, D.; Theis, M.; Pakula, T.; Wegner, G.; Matyjaszewski, K. Macromolecules 2006, 39, 584-593.
(31) Lee, H.; Jakubowski, W.; Matyjaszewski, K.; Yu, S.; Sheiko, S. S. Macromolecules 2006, 39, 4983-4989.
(32) Yu-Su, S. Y.; Sheiko, S. S.; Lee, H.-i.; Jakubowski, W.; Nese, A.; Matyjaszewski, K.; Anokhin, D.; Ivanov, D. A. Macromolecules 2009, 42, 9008-9017.
(33) Li, C.; Gunari, N.; Fischer, K.; Janshoff, A.; Schmidt, M. Angew. Chem. Int. Ed., 2004, 43, 1101 -1104.
(34) Pietrasik, J.; Sumerlin, B. S.; Lee, R. Y.; Matyjaszewski, K. Macromolecular Chemistry and Physics 2007, 208, 30-36.
(35) Yamamoto, S.-i.; Pietrasik, J.; Matyjaszewski, K. Macromolecules 2007, 40, 9348-9353.
(36) Lee, H.-i.; Boyce, J. R.; Nese, A.; Sheiko, S. S.; Matyjaszewski, K. Polymer 2008, 49, 5490-5496.
(37) Nese, A.; Lebedeva, N. V.; Sherwood, G.; Averick, S.; Li, Y.; Gao, H.; Peteanu, L.; Sheiko, S. S.; Matyjaszewski, K. Macromolecules 2011, 44, 5905–5910.
(38) Yamamoto, S.-i.; Pietrasik, J.; Matyjaszewski, K. Macromolecules 2008, 41, 7013-7020.
(39) Sitti, M.; Cusick, B.; Aksak, B.; Nese, A.; Lee, H.-i.; Dong, H.; Kowalewski, T.; Matyjaszewski, K. ACS Appl. Mater. Interfaces 2009, 1, 2277-2287.
(40) Lee, H.-i.; Pietrasik, J.; Sheiko, S. S.; Matyjaszewski, K. Prog. Polym. Sci. 2010, 35, 24-44.