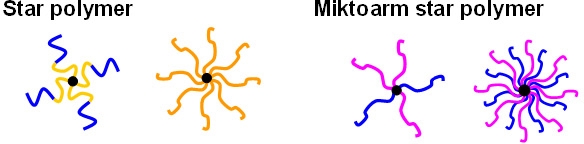
Star Copolymers
Core first
Arm first
Mikto-arm star copolymer
Is CRP required?
Star Copolymers
Introduction:
Star polymers consist of several linear polymer chains connected at one point.(1) Star molecules prepared by anionic polymerization had been examined prior to the development of CRP. However due to the limitations of ionic polymerization the composition and functionality of the materials were limited but their compact structure and globular shape provide them with a set of unique properties, such as low solution viscosity, and the core shell architecture enabled several potential applications spanning a range from thermoplastic elastomer's(2) to drug carriers.(3)
Based on the chemical compositions of the arm species, star polymers can be classified into two categories: homo-arm (or regular) star polymers or mikto-arm (or heteroarm) star copolymers. Homo-arm star polymers consist of a symmetrical structure comprising radiating arms with identical chemical composition and similar molecular weight. In contrast, a miktoarm star molecule contains two or more arm species with different chemical compositions and/or molecular weights and/or different peripheral functionality. There are several approaches that can be employed for synthesis of star copolymers.
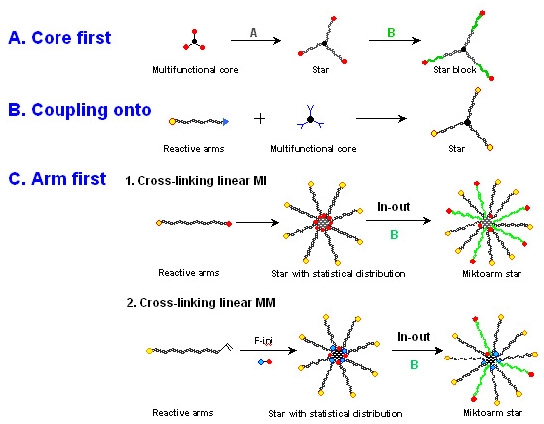
As suggested above, star polymers can be synthesized by variations on one of three methods:
- The "core-first" approach, where the controlled polymerization is conducted from either a well defined initiator with a known number of initiating groups(4-9) or a less well defined multifunctional macromolecule.(10-13)
- An approach that until recently had not received as much attention is the "coupling onto" approach where a tele-functional linear molecule is reacted with a preformed core molecule containing complementary functionality.(14-17) In order to improve the coupling efficiency, a highly efficient organic coupling reaction required, such as click reactions.(18,19) The preparation of stars using click chemistry could be applied to almost any of the strategies discussed on this page.(14)
- There are two approaches to the "arm-first" synthesis of star polymers.(20-24) One is where a linear "living" copolymer chain, or added macroinitiator, is linked by continuing the copolymerization of the mono-functional macroinitiator with a divinyl monomer forming a crosslinked core.
- The other, is the direct copolymerization of a macromonomer with a divinyl monomer in the presence of a low molecular weight initiator.(25,26)
- A combination of "arm-first" and "core-first" methods is particularly useful for synthesis of miktoarm star copolymers. One employs the retained initiating functionality in the formed "arm first" core to initiate the polymerization of a second monomer in a "grafting out" or a "grafting from" copolymerization.(27-31)
Core first:
The following section on nano-composites formed by grafting from the surface of a functionalized particle is an evolution of the "core first" approach to synthesis of star macromolecules. Initially a low molecular weight multi-functional molecule was used in a "grafting from" reaction to form star macromolecules with a well defined number of arms. Use of hexakis(chloromethyl)benzene as a well defined multifunctional initiator for the polymerization of styrene provided the first multi-arm polymer prepared using ATRP.(5,7) The composition of the core and arms were quickly expanded.(7-9,32)
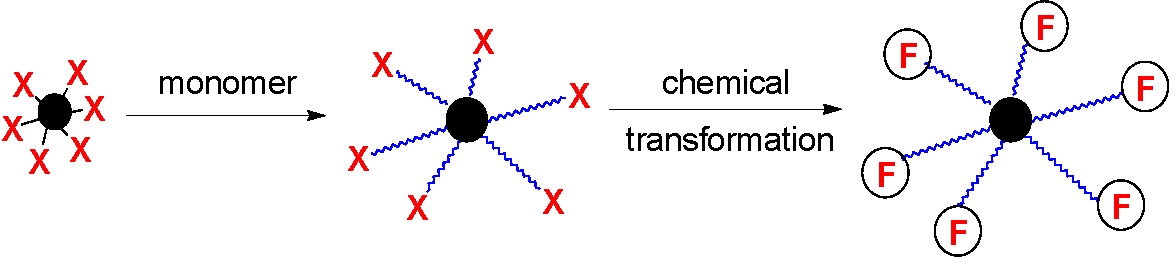
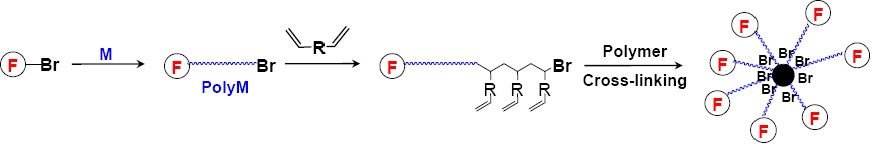
Since the tethered chains in a grafting from reaction retain their terminal functionality they could be chain extended to form star block copolymers and/or the radically transferable atoms on the chain ends (X) could be converted to other functional groups (F) suitable for post-polymerization functionalization reactions.(8)
Less well-defined multi arm star structures were prepared by grafting from a soluble multifunctional hyperbranched core. The core was prepared by polymerizing a molecule containing both a reactive group suitable to initiate a CRP and a polymerizable double bond, an initiator/monomer or inimer. Self-condensing vinyl polymerization using ATRP has been applied to inimers, i.e. monomers that also contain activated halogen atoms, such as α-bromoesters and benzyl halides.(11-13) For example, when 2-bromopropionyloxyethyl acrylate was polymerized in the presence of a copper catalyst with 4,4'-dinonyl-2,2'-bipyridine ligands, a hyperbranched polymer with degree of branching ~0.5 and DP = 78 was obtained. Each soluble hyperbranched polymer contained on average 78 active bromopropionyloxy groups which were used to initiate the polymerization of methyl acrylate forming a star with 78 arms.
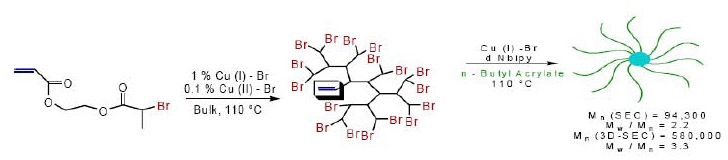
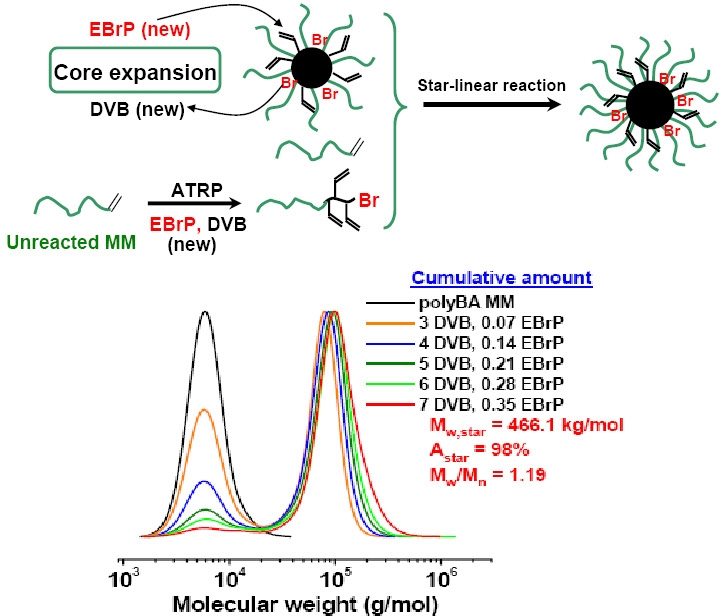
A simple sequential polymerization of a crosslinker followed by polymerization of a monomer(33) provides a broadly applicable approach to star copolymers. This novel method, termed "star from in situ generated core", belongs to the broader category of "core-first" methodology and presents an alternative strategy for star synthesis when compared to the traditional "arm-first" method, in which monomer is polymerized first followed by formation of the core by (co)polymerization of a cross-linker.
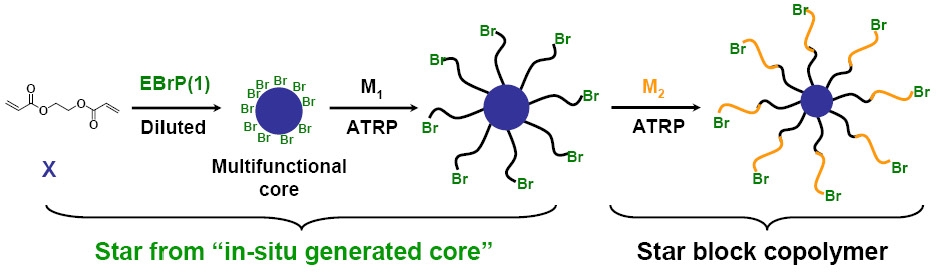
To illustrate this new concept for the synthesis of star polymers using controlled radical polymerization techniques, ATRP was applied to the homopolymerization of a crosslinker, ethylene glycol diacrylate (EGDA) to generate a multifunctional crosslinked core (nanogel). Monovinyl monomers were added to the reaction mixture at high conversion of the crosslinker and were polymerized from the polyEGDA nanogel macroinitiator (MI) to form the star arms. A spectrum of different acrylate monomers were polymerized from the first formed core providing star polymers with different compositions for the arms.
Several parameters affected the structure of the star, such as the initial concentration of the crosslinker, the molar ratio of crosslinker to initiator, and the moment of addition of the monovinyl monomer to the ongoing polymerization reaction. The star polymers preserved the initiating sites at the chain ends, and they were further used as star MIs for arm extension by polymerization of a second monovinyl monomer to form a star block copolymer.
Indeed this approach of preparing well defined polymers by copolymerization of a crosslinker and a monomer can be expanded to provide access to a full range of stars/gels/networks.(16)
Arm first
Macroinitiator approach:
The "arm first" approach forms the core of the star macromolecule by coupling monofunctional "living" polymeric chains with a difunctional reagent and was first applied to living anionic polymerization.(20) A similar approach was also successful using ATRP.(21,22) Initially the simple chain extension of a linear macroinitiator with a crosslinker provided star macromolecules with broad polydispersity as a result of star-star coupling reactions.(21)
There are several parameters in an ATRP that should be carefully controlled in order to maximize the yield of stars and prevent/reduce star-star coupling reactions. Some detailed studies have been carried out on the coupling of monofunctional polystyrenes and polyacrylates with divinylbenzene (DVB) and di(meth)acrylates to prepare star polymers and the following guidelines were developed:(23-26)
- The ratio of di-functional reagent to growing chains seems to be optimal in the range of 10-20.
- Monomer conversion (or reaction time) has to be carefully controlled and stopped before star-star coupling occurs. It seems that ~5% of arms can not be incorporated into the star macromolecules under typical one step conditions.
- Higher yields of stars are observed for polyacrylates than for polystyrenes. This may be attributed to a higher proportion of terminated chains (due to slower propagation and higher concentration of radicals) in styrene polymerization under "standard" ATRP conditions. (The use of ARGET ATRP for the synthesis of the active growing polymer chains should change both these observations.)
- The choice of the difunctional reagent is important and reactivity should be similar to, or lower than that of the arm-building monomers.
- Halogen exchange slightly improves the efficiency of star formation.
- Solvent, temperature and catalyst concentration should be also optimized.
Under typical conditions, ~50 arms are coupled into star structures. Apparent molecular weights, measured by GPC, show a 10-20-fold increase of molecular weight (from Mn = 10,000 to Mn ~ 150,000) but light scattering indicates that the stars have a much higher molecular weight (Mn ~500,000).
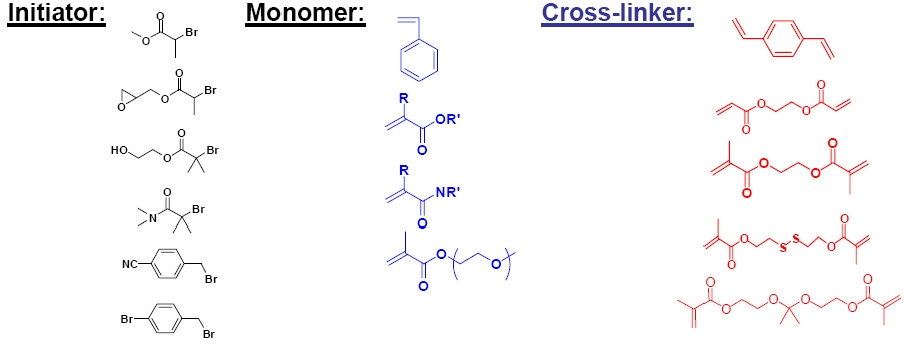
A range of initiators, monomers and cross linkers was examined for the preparation of star molecules with peripheral functionality and cores of differing phobicity.
Another approach to form star polymers in high yield was via cross-linking self organized amphiphilic macroinitiators by AGET ATRP in aqueous dispersed media.(34) Linear poly(ethylene oxide)-b-polystyrene (PEO-PS-Cl) block copolymers were the arm precursors. The amphiphilic block copolymers with halogen chain-end functionality formed divinyl cross-linker swollen micelles in water and then were cross-linked by the cross-linkers when polymerization was initiated. Due to the formation of micelles before the polymerization was initiated, star-star or star-linear chain reactions were not required for the star formation. This suppressed star-star coupling reactions and resulted in the formation of star polymers with low PDI (Mw/Mn < 1.1) but still high molecular weight (over 1000 kg/mol).
Macromonomer Approach:
Another approach to arm first star copolymers, with available core containing initiating functionality, is the CRP of higher molecular weight macromonomers in a pure homo-polymerization(26) initiated with a small molecule initiator which usually leads to a "brush molecule" with a degree of polymerization of 10-25, which from a topological standpoint can be considered a star.
An alternate approach is a copolymerization of macromonomers with a divinyl crosslinking monomer.(25,27)
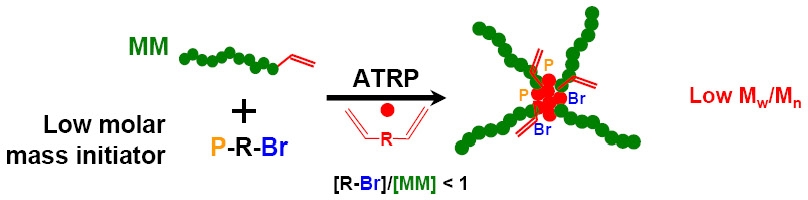
The sequential addition of additional initiator and crosslinker to the reaction increases the number of macromonomer units incorporated into each star.(35)
A potential application for homo-arm stars was illustrated by preparation of two well defined star macromolecules with two oppositely charged arm structures; a poly[2-(dimethylamino)ethyl methacrylate] (PDMAEMA) star and a poly(acrylic acid) (PAA) star with cross-linked cores.(36) Exploitation of the electrostatic interactions between the polyelectrolyte arms of PDMAEMA star and PAA star polymers generated alternating polyelectrolyte multilayer LbL films using layer-by-layer (LbL) assembly.
Mikto-arm star copolymers
In-out Method
Both approaches to form star molecules using the arm first approach retain the initiating functionality within the core of the star and therefore provide a simple approach to form mikto-arm stars by conducting a controlled polymerization of a second monomer from the accessible initiator functionality in the core.
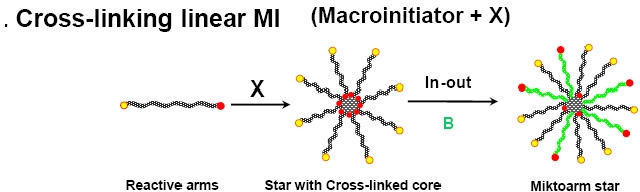
As noted above one arm-first procedure involves the synthesis of a linear mono-functional polymer chain polyA, which is used as a macroinitiator (MI) in a subsequent cross-linking reaction using a divinyl compound to produce a (polyA)n-polyX star polymer, where polyX represents the core of the star polymer and n is the number of polyA arms. The initiating sites are preserved within the core of the star polymer (i.e., alkyl halide groups in ATRP) and the (polyA)n-polyX star polymer can be used as a multifunctional star initiator in a chain extension reaction with a different monomer, B, to yield a miktoarm star copolymer, (polyA)n-polyX-(polyB)m. This combination method for synthesizing miktoarm star copolymers was termed the "in-out" method.(28,30)
The efficiency of initiation of the second set of arms is dependent on the compactness of the first formed core with less densely crosslinked cores providing more efficient initiation i.e., a greater fraction of the encapsulated initiator sites, for the grafting out polymerization.(31)
Normally, one seeks to, or has to, form a chemically stable core. However, it is possible to select a crosslinking agent with a degradable link between the two functional crosslinking groups and prepare a material with a degradable core.
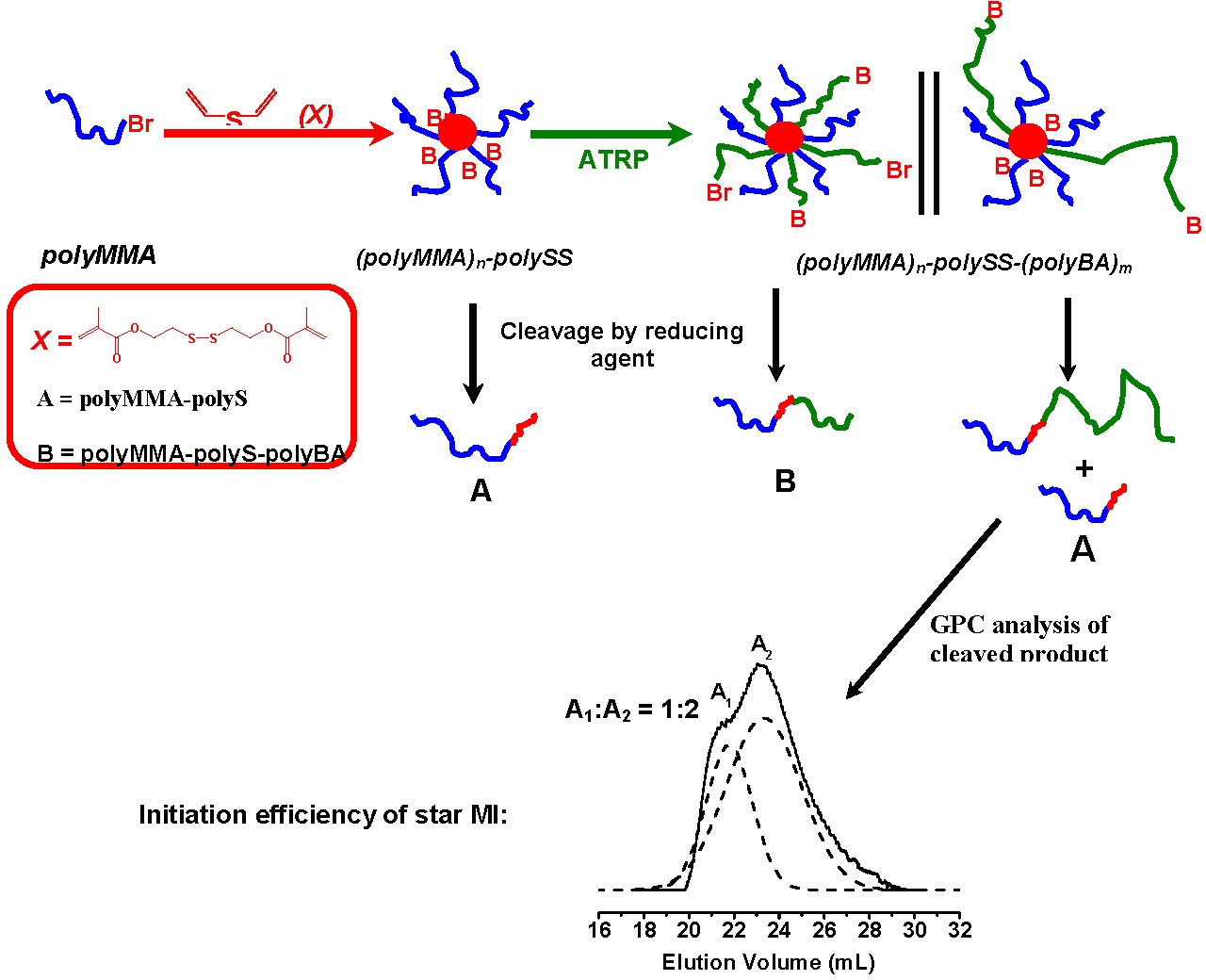
This was accomplished by linking the first formed arms with a dimethacrylate crosslinker containing a disulfide link between the methacrylate units. As shown in the above scheme the mikto-arm star copolymer could be degraded in a reducing environment to form a mixture of an AB block copolymer and some residual A-homopolymer. The ratio between the block copolymer and homopolymer gave a direct measurement of the initiating efficiency of the constrained core initiating units; in this example it was only ~20%.(30)
We recently reported a new strategy for synthesis of miktoarm star copolymers using a simple and general "arm-first" method, i.e., one-pot ATRP cross-linking a mixture of different linear MIs and/or MMs with a divinyl cross-linker in order to synthesize miktoarm star copolymers with potentially any desired molar ratio and composition of the arms.(35,37)
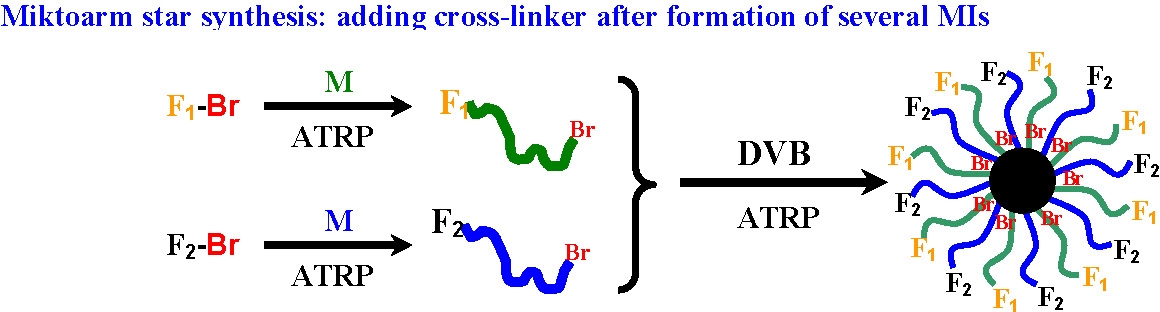
Liquid chromatography under the critical conditions (LCCC) for each of the homopolymers arms provided proof that the star structure a miktoarm star copolymer containing two or more compositionally different arm species in one molecule, and was not a mixture of different homo-arm star polymers.(35) Miktoarm star copolymers containing five kinds of arms were synthesized for the first time by copolymerizing a mixture of five linear MIs with different chemical composition. When linear MMs were partially or completely used as arm precursors instead of MIs, miktoarm star copolymers with high star yield and low polydispersity were successfully synthesized.(35)
A recent example of a combination of an "arm first" and a "core first" approach is "grafting onto" a functionalized core via "click" chemistry.(14,15) Three-arm and four-arm star polystyrene polymers were synthesized by a combination of ATRP and click coupling chemistry. The click reaction between an azido-terminated polystyrene (PS-N3) and an alkyne-containing multifunctional compound proved to be fast and efficient. When an azido-terminated polystyrene polymer was reacted with a trialkyne-containing or tetraalkyne-containing compound, the yields of 3-arm star and 4-arm star polymers were around 90% and 83%, respectively.
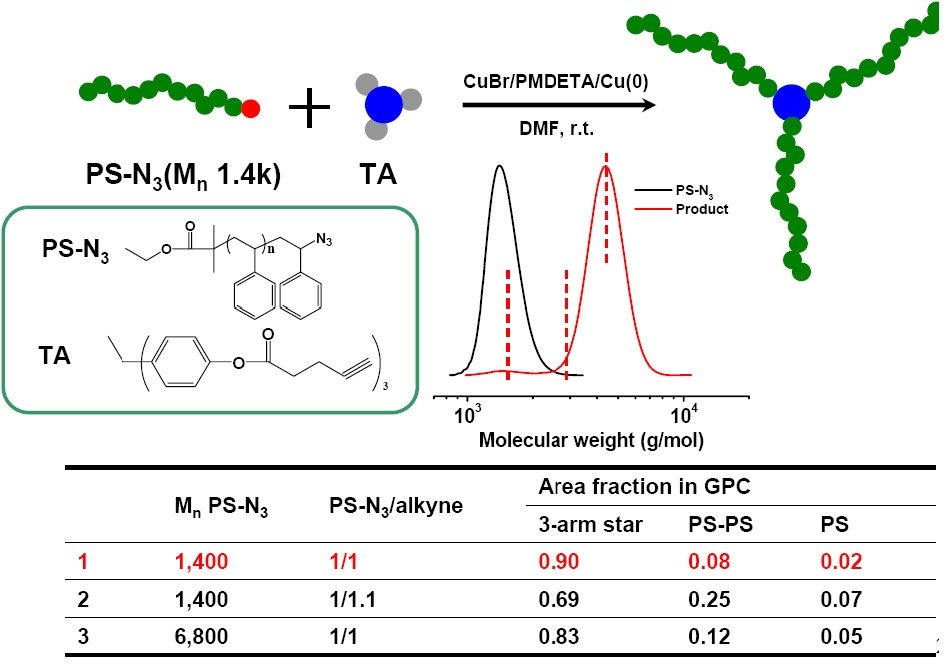
The influence of several parameters on the efficiency of the click coupling reaction was studied, including the molecular weight of the PS-N3 polymer, the presence of an added reducing agent, Cu0, and the stoichiometry between the azido and alkynyl groups. The results indicated that the yield of the coupled product was higher when a lower molecular weight PS-N3 was employed in conjunction with a small amount of reducing agent, and the molar ratio of azido and alkynyl groups was close to 1.
A final question for this section on preparation of star macromolecules could be "Is a CRP required for preparation of star copolymers"?
Obviously the question would not be asked in a public forum if the answer was yes. It was recently disclosed how a standard free radical polymerization can be used for the high-yield synthesis of uniform star copolymers.(38) In the paper a novel simple method was reported for the synthesis of well-defined star polymers in high star yield by applying a conventional radical polymerization procedure to the copolymerization of linear macromonomers and divinyl cross-linkers in a homogeneous solution of the reagents.
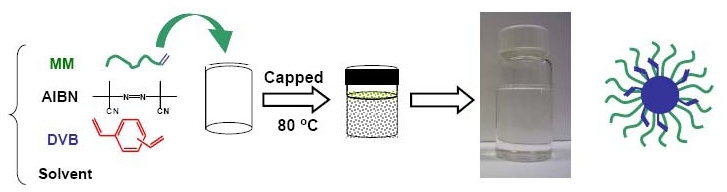
The advantages of conventional radical polymerization for star synthesis, as compared to other polymerization techniques, include but are not limit to:
- no rigorous deoxygenation process is required,
- no costly synthesis of a radical mediating agent and
- no catalyst contamination in the final product.
The efficiency of star polymer formation (yield and polydispersity) depends on selection of appropriate conditions. Star growth continued beyond the time required for decomposition of the initiator as radicals were trapped within the congested core thereby reducing star -star coupling reactions and multi-arm star macromolecules with narrow polydispersity could be prepared.
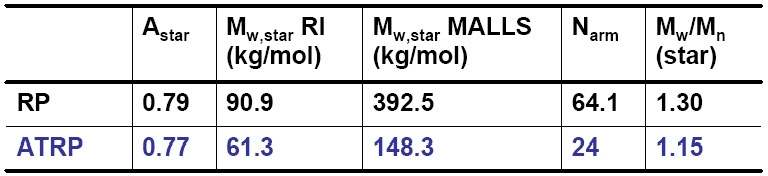
Use of macromonomers with α-chain end functionality (not the vinyl group) allows synthesis of stars with a shell of peripheral functionality.
In summary star molecules can be prepared in high star yield with low polydispersity and site specific functionality. They can be prepared to be biocompatible with high transfection efficiency.
REFERENCES
(1) Hawker, C. J. Angew. Chem., Int. Ed. Engl. 1995, 34, 1456-1459.
(2) Quirk, R. P.; Kim, J. Rubber Chemistry and Technology 1991, 64, 450-468.
(3) Hadjichristidis, N.; Pitsikalis, M.; Pispas, S.; Iatrou, H. Chemical Reviews 2001, 101, 3747-3792.
(4) Li, M.; Jahed, N. M.; Min, K.; Matyjaszewski, K. Macromolecules 2004, 37, 2434-2441.
(5) Wang, J.-S.; Greszta, D.; Matyjaszewski, K. Polym. Mater. Sci. Eng. 1995, 73, 416-417.
(6) Matyjaszewski, K.; Wang, J.-S. In PCT Int. Appl.; (Carnegie Mellon University, USA). WO 9630421, 1996; p 129 pp.
(7) Matyjaszewski, K.; Miller, P. J.; Pyun, J.; Kickelbick, G.; Diamanti, S. Macromolecules 1999, 32, 6526-6535.
(8) Angot, S.; Murthy, K. S.; Taton, D.; Gnanou, Y. Macromolecules 1998, 31, 7218-7225.
(9) Ueda, J.; Kamigaito, M.; Sawamoto, M. Macromolecules 1998, 31, 6762-6768.
(10) Gaynor, S. G.; Edelman, S.; Matyjaszewski, K. Macromolecules 1996, 29, 1079-1081.
(11) Matyjaszewski, K.; Gaynor, S. G.; Kulfan, A.; Podwika, M. Macromolecules 1997, 30, 5192-5194.
(12) Matyjaszewski, K.; Gaynor, S. G.; Mueller, A. H. E. Macromolecules 1997, 30, 7034-7041.
(13) Matyjaszewski, K.; Gaynor, S. G. Macromolecules 1997, 30, 7042-7049.
(14) Gao, H.; Matyjaszewski, K. Macromolecules 2006, 39, 4960-4965.
(15) Gao, H.; Min, K.; Matyjaszewski, K. Macromolecular Chemistry and Physics 2007, 208, 1370-1378.
(16) Gao, H.; Matyjaszewski, K. Progress in Polymer Science 2009, 34, 317-350.
(17) Inglis, A. J.; Sinnwell, S.; Davis, T. P.; Barner-Kowollik, C.; Stenzel, M. H. Macromolecules 2008, 41, 4120-4126.
(18) Kolb, H. C.; Finn, M. G.; Sharpless, K. B. Angewandte Chemie, International Edition 2001, 40, 2004-2021.
(19) Sumerlin, B. S.; Tsarevsky, N. V.; Louche, G.; Lee, R. Y.; Matyjaszewski, K. Macromolecules 2005, 38, 7540-7545.
(20) Rein, D.; Lamps, J. P.; Rempp, P.; Lutz, P.; Papanagopoulos, D.; Tsitsilianis, C. Acta Polymerica 1993, 44, 225-229.
(21) Xia, J.; Zhang, X.; Matyjaszewski, K. Macromolecules 1999, 32, 4482-4484.
(22) Zhang, X.; Xia, J.; Matyjaszewski, K. Macromolecules 2000, 33, 2340-2345.
(23) Baek, K.-Y.; Kamigaito, M.; Sawamoto, M. Macromolecules 2001, 34, 7629-7635.
(24) Gao, H.; Matyjaszewski, K. Macromolecules 2006, 39, 3154-3160.
(25) Gao, H.; Ohno, S.; Matyjaszewski, K. Journal of the American Chemical Society 2006, 128, 15111-15113.
(26) Gao, H.; Matyjaszewski, K. Macromolecules 2007, 40, 399-401.
(27) Hadjichristidis, N. J. Polym. Sci., Part A: Polym. Chem. 1999, 37, 857-871.
(28) Georgiades, S. N.; Vamvakaki, M.; Patrickios, C. S. Macromolecules 2002, 35, 4903-4911.
(29) Du, J.; Chen, Y. Macromolecules 2004, 37, 3588-3594.
(30) Gao, H.; Tsarevsky, N. V.; Matyjaszewski, K. Macromolecules 2005, 38, 5995-6004.
(31) Gao, H.; Matyjaszewski, K. Macromolecules 2006, 39, 7216-7223.
(32) Matyjaszewski, K. Polymer International 2003, 52, 1559-1565.
(33) Gao, H.; Matyjaszewski, K. Macromolecules 2008, 41, 1118-1125.
(34) Li, W.; Matyjaszewski, K. J. Am. Chem. Soc. 2009, 131, 10378-10379.
(35) Gao, H.; Matyjaszewski, K. Macromolecules 2008, 41, 4250-4257.
(36) Kim, B.-S.; Gao, H.; Argun, A. A.; Matyjaszewski, K.; Hammond, P. T. Macromolecules 2009, 42, 368-375.
(37) Gao, H.; Matyjaszewski, K. Journal of the American Chemical Society 2007, 129, 11828-11834.
(38) Gao, H.; Matyjaszewski, K. Chem.--Eur. J. 2009, 15, 6107-6111.