Structural Characterization of an ATRP Catalyst Complex
Structure of a Catalyst Complex
Effect of Substituents on the Ligands:
Structure of a Catalyst Complex
The primary roles of the ligand in an ATRP catalyst complex is to solubilize the transition metal salts in the polymerization medium and to adjust the redox potential of the metal center to provide appropriate activity and dynamics for the repetitive halogen exchange reaction. The electron donating ability of the ligand can greatly affect the redox potential of the transition metal complex and influence the reactivity of the metal center in halogen abstraction and transfer reactions. This means that the structure of the catalyst selected for a given reaction affects the kinetics of an ATRP and hence the degree of control over the polymerization reaction under a selected set of polymerization conditions.
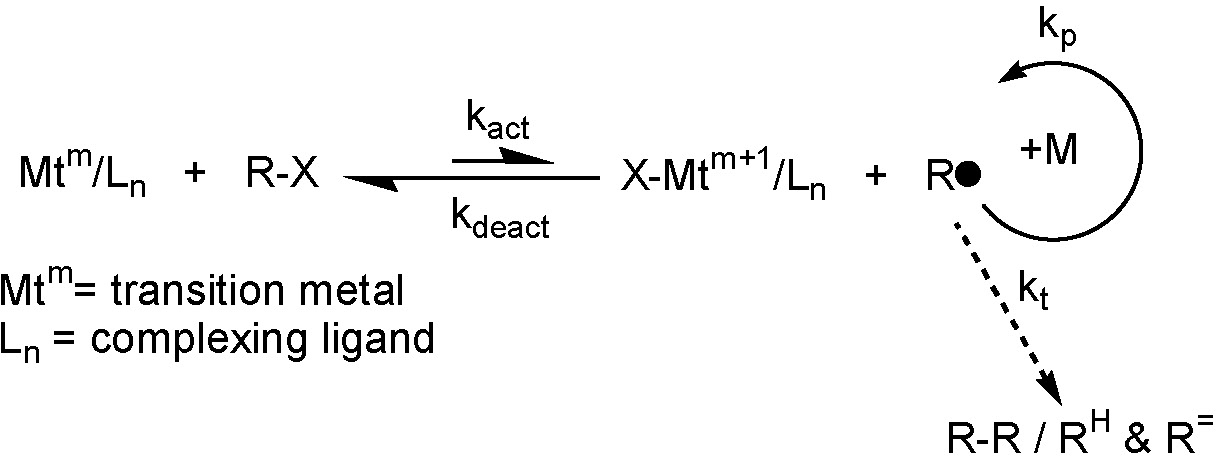
In the above scheme, representing an ATRP equilibrium, Mt is a transition metal with two stable oxidation states differing by 1 (m <-> m+1). As noted elsewhere on this web site the bulk of the work on ATRP conducted by the Matyjaszewski group has employed copper as the transition metal. Copper has provided numerous catalysts with a range of nitrogen containing ligands that have been shown to be suitable for controlled polymerization of a broad range of radically copolymerizable monomers. Therefore, structural characterization of a series ATRP active copperI and copperII complexes with a spectrum of ligands continues to be studied within the Matyjaszewski group using a variety of analytical tools.
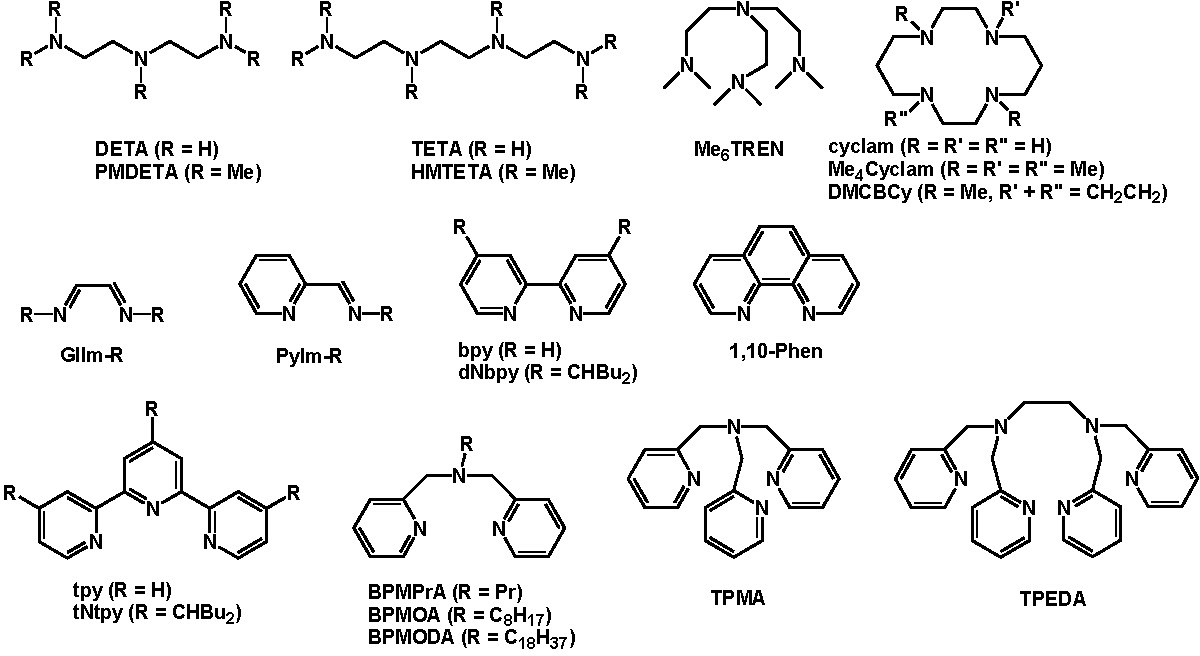
Recent reviews on the structural aspects of copper catalyzed ATRP (1-6) provide some background on the fundamentals of transition metal catalyzed atom transfer reactions, including ATRA and ATRP. The reviews focus on the structure of a catalyst complex formed with bidentate, tridentate and tetradentate nitrogen ligands that generally work well for Cu-mediated ATRP. The choice of ligand greatly influences the effectiveness of the catalyst in a specific polymerization reaction. Since catalyst activity now spans nine orders of magnitude one ligand does not work for every copolymerization under a “standard” set of conditions. Within the review articles a broad series of ligands forming catalyst complexes with an expansive range of activity are discussed including:
2,2'-bipyridine (bpy),
4,4'-di(5-nonyl)-2,2'-bipyridine (dNbpy),
N,N,N',N'-tetramethylethylenediamine (TMEDA),
N-propyl(2-pyridyl)methanimine (NPrPMI),
2,2':6',2''-terpyridine (tpy),
4,4',4''-tris(5-nonyl)- 2,2':6',2''-terpyridine (tNtpy),
N,N,N',N'',N''-pentamethyldiethylenetriamine (PMDETA),
N,N-bis(2-pyridylmethyl)octylamine (BPMOA),
1,1,4,7,10,10-hexamethyltriethylenetetramine (HMTETA),
tris[2-(dimethylamino)ethyl]amine (Me6TREN),
tris[(2-pyridyl)methyl]amine (TPMA),
1,4,8,11-tetraaza-1,4,8,11-tetramethylcyclotetradecane (Me4CYCLAM) and
N,N,N',N'-tetrakis(2-pyridylmethyl)ethylenediamine (TPEN)
The structures of copperI and copperII complexes with the following ligands are discussed in the context of how structure affects catalyst activity in addition to solvent and temperature.
diethylenetriamine (DETA),
triethylenetetramine (TETA),
N,N-bis(2-pyridylmethyl)amine (BPMA),
tris[2-aminoethyl]amine (TREN) and
1,4,8,11-tetraazacyclotetradecane (CYCLAM)
N,N,N',N'-tetrakis(2-pyridylmethyl)ethylenediamine (TPEN)
The ligands most frequently employed for copper based catalysts are presented below:
Aspects of the structural studies concentrate on the stoichiometry between the complex forming ligand and the copper centers,(7, 8) determination of the geometry of the formed complexes, and their solution behavior. (9-11) Techniques used include solid state X-ray crystallography, Extended X-ray Absorption Fine Structure (EXAFS), (9) Electrospray Ionization Mass Spectrometry (ESI-MS), (11) and UV-Vis, Raman and Far IR spectroscopy.
A series of Cu(II) complexes were examined and they adopted either a trigonal bipyramidal structure, as in the case of the dNbpy ligand, or a distorted square pyramidal coordination, in the case of triamines and tetramines. (12)
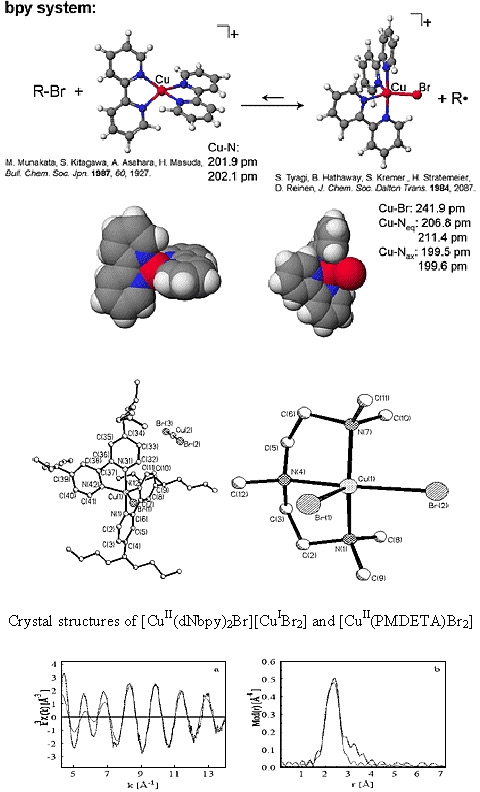
Depending on the type of amine ligand, the complexes were either neutral (triamines) or ionic (bpy and tetramines). The counterions in the case of the ionic complexes were either a bromide anion (Me4Cyclam and HMTETA) or the linear [CuIBr2]- anion (dNbpy).
No direct correlation was found between the length of the CuII-Br bond and the deactivation rate constant in an ATRP, which suggests that other parameters such as the entropy for the structural reorganization between the CuI and CuII complexes might play an important role in determining the overall activity of the catalyst in ATRP.(5)
Effect of Substituents on the Ligands:
Since the first employed CuX(bpy)2 catalytic system was heterogeneous in non-polar media early work on incorporation of substituents into ligand for an ATRP focused on controlling the solubility of the catalyst complex. Incorporation of alkyl-substituents in the bpy ligand provided soluble catalyst complexes and polymers with low dispersity and a means to determine the kinetics of the reaction. (13, 14) 4,4'-Di-n-heptyl-2,2'-bipyridine (dHbpy), 4,4'-di(5-nonyl)-2,2'-bipyridine (dNbpy) and 4,4'-di-5-nonyl-2,2'-bipyridine (bpy9) were synthesized to provide homogeneous copper complexes in ATRP. (13, 15) However, as noted above, in addition to modifying the solubility of the catalyst complex the electron donating ability of the ligands greatly affect the redox potential of the transition metal complex and influence the reactivity of the metal center in halogen abstraction and transfer. (16) Catalysts therefore provide synthetic freedom in ligand design to manipulate and tune catalytic properties, which had not been truly exploited in ATRP.
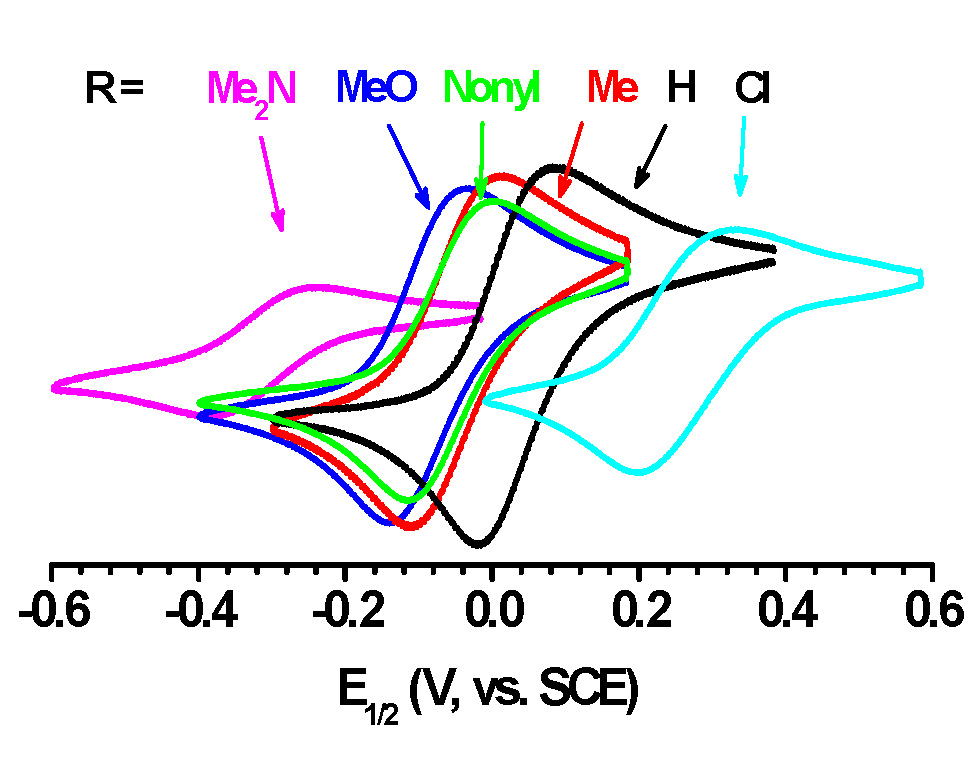
Recent work has systematically examined the effect of incorporating electron donor groups (EDG) into potential ATRP ligands. (17) This initial paper focused on examining the importance of electronic effects when employing various para substituents (R) in bpy ligands. The substituents ranged from electron withdrawing groups (EWGs: Cl) to electron donating groups (EDGs: Me-, MeO-, (Me)2N-), and represented the first systematic study seeking to correlate structure-activity of substituents bearing variable Hammett parameters (σρ) to E1/2 (KATRP) and polymerization rate, see following table.
Table 1: CV of 4,4'-Substituted Bipyridyl Ligands.
|
Entry |
R |
σρa |
E1/2 (V)b |
Δ Ep (mV) |
|
1 |
Cl |
0.23 |
0.270 |
130 |
|
2 |
H |
0 |
0.055 |
110 |
|
3 |
Me |
-0.17 |
-0.048 |
125 |
|
4 |
dN |
n/a |
-0.055 |
120 |
|
5 |
MeO |
-0.27 |
-0.088 |
105 |
|
6 |
(Me)2N |
-0.83 |
-0.313 |
145 |
aLiterature values,(16) bV vs. Saturated Calomel Electrode (SCE)
It was expected that electron withdrawing groups (EWG) would stabilize CuI while electron donating groups (EDG) would stabilize CuII and indeed when values for E1/2 are considered, changing the substituents significantly changed the activity of the catalyst complexes; ∆ (-Cl to -NMe2) ≈ 600 mV, The only Bpy ligand evaluated in the study that is currently not commercially available is the most active (Me)2N-Bpy. Changing from -H to -NMe2 provides a difference in KATRP of one million, see Figure B, making the p-(Me)2N-Bpy similar in activity to one of the more active complexes in ATRP, namely Cu/Me6TREN. Indeed as discussed in the paper (17) the (Me)2N-Bpy ligand is so active that it could not be used in a standard ATRP but only in ATRP procedures with low ppm levels of catalyst.
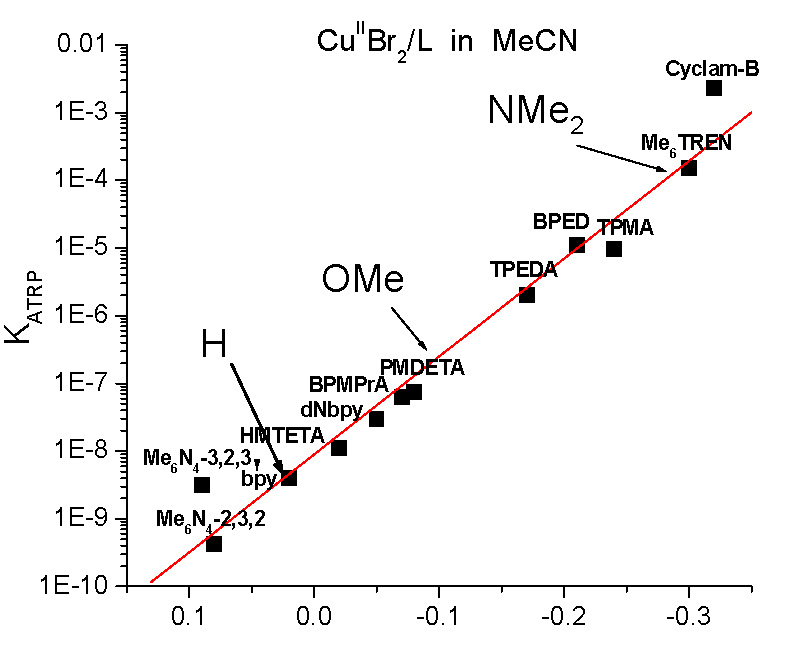
Control in ATRP is related to both the efficiency of radical deactivation, kd, but also the concentration of deactivator, [CuII/L]. Since more active catalysts have larger values of KATRP, this inherently leads to a greater fraction of CuII/L deactivator in the reaction medium. This means, theoretically, that with higher activity catalysts a lower concentrations of total copper can be expected to provide good control over both molecular weights and molecular weight distributions. Therefore the highly active R-bpy ligands were investigated to determine their ability to maintain CLRP behavior with ppm concentrations of catalyst. ATRP conducted with 500 ppm of CuII showed that increasing electron donating character of the substituents resulted in higher rates of polymerizations and polymers of similar Mn had lower Mw/Mn values when prepared using more active catalysts.
Catalyst development plays a pivotal role in the endeavor to reduce catalyst loadings, overcome limitations in monomer selection and achieve high levels of control in ATRP systems. Based on the rational design, introduced above, the currently most active series of copper catalyst for atom transfer radical polymerization (ATRP) was prepared based on the tris[(2-pyridyl)methyl]amine (TPMA) ligand.(19) The performance of these ATRP catalysts was evaluated by cyclic voltammetry, stopped-flow measurements, and simulations. The new ligands, containing up to nine EDGs in each ligand, (named TPMA*1, TPMA*2 and TPMA*3 for the number of tri-substituted pyridine rings in the ligand) were employed to form catalysts that were evaluated in various ATRP methods, including normal ATRP, ARGET, ICAR or eATRP, which provided important insights into how to use very active catalysts in ATRP.
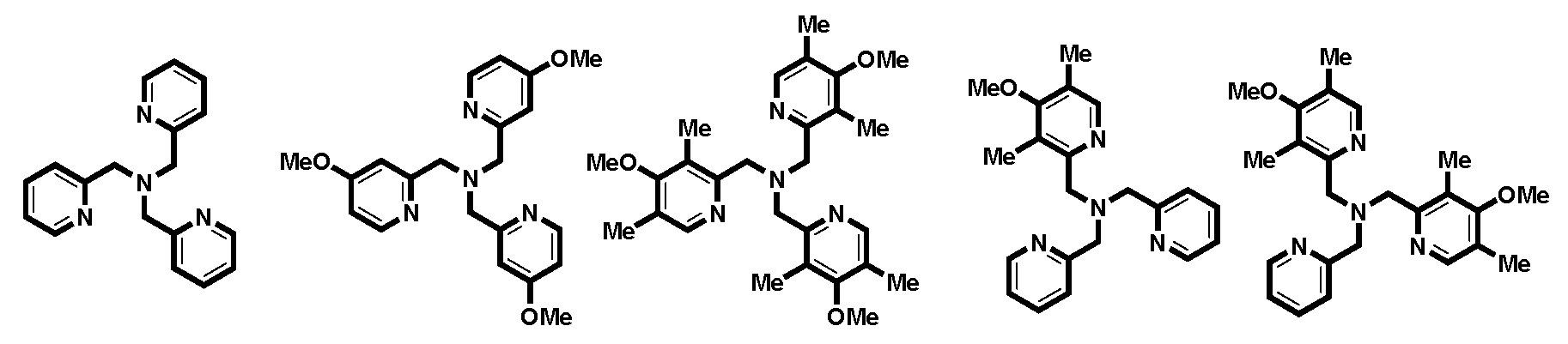
TPMA TPMA-OMe TPMA*3 TPMA*1 TPMA*2
Counter intuitively, the standard ATRP reaction was slow with the ligand containing all the activating EDGs, only low conversions and moderate degrees of control were observed. After 1 h, the monomer conversion was 7% and the resulting polymers had a dispersity, Mw/Mn = 1.34. At longer reaction times no significant conversion increase was achieved, i.e. after 5.15 hours, conversion was only 13.5 % with no significant increase in conversion at longer reaction times, while providing polymer with moderately broad dispersity, 1.25-1.35.
This behaviour can be attributed to the highly active nature of CuI/TPMA* catalyst complex. Due to the very fast rates of activation, very active catalysts generate an initially high concentration of radicals which results in a significant loss of chain-end functionality. Without the living (macro)alkyl halide, polymerization cannot occur under “normal” ATRP conditions. An ARGET ATRP with 50 ppm X-CuII/TPMA* catalyst, activated by reaction with different mole ratio’s of Sn(EH)2, provided linear first order reaction kinetics and reached 90% conversion with dispersity 1.1. Similar results were obtained with e(ATRP), SARA ATRP and ICAR ATRP reactions with the catalyst complexes containing the EDGs providing better control than with the “old” very active TPMA ligand. The catalyst complex formed with TPMA* is three orders of magnitude more active than the complex formed with TPMA and provided a well-controlled reaction with only 5 ppm catalyst.
After this development of the most active ATRP catalyst with a tris[((4-methoxy-2,5-dimethyl)-2-pyridyl)methyl]amine (TPMA*3) ligand, which has three electron-donating groups on each pyridine ring (17, 19) and led to a catalyst that is 5 million times more active than the seminal catalytic system employing the 2,2-bipyridine (bpy) ligand and 1000 times more active than the commonly used tris(pyridylmethyl)amine TPMA) ligand, illustrated in the following figure. (5)
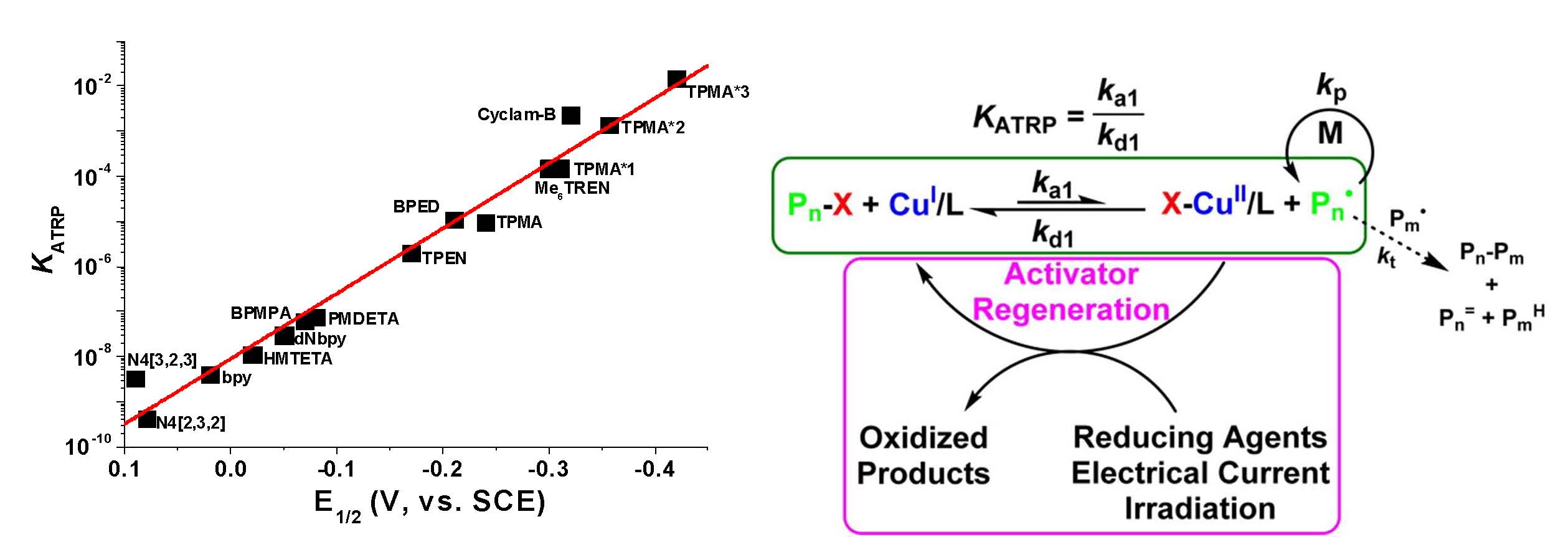
The schematic on the right hand side of the figure indicates that these very active catalyst complexes are best employed in low catalyst regeneration procedures: SARA ATRP, ICAR ATRP, ARGET ATRP, eATRP, photoATRP, mechanoATRP, for polymerization of styrenes, (meth)acrylates, acrylonitrile, acidic monomers at catalyst loadings < 100 ppm providing access to copolymers with many different architectures, including stars, brushes, block copolymers. This is because ATRP systems with activator regeneration follow steady-state kinetics for which the [CuII]/[CuI] ratio is dynamic and depends on the instantaneous [R*] and [RX] concentration and KATRP. “Normal” ATRP on the other hand follows the persistent radical effect and does not involve a dynamic equilibrium.
According to Hammett parameters, using the even more electron-donating dimethyl amino (−NMe2) group should further increase the catalyst activity, as already observed for bpy derivatives. (17) Therefore, we considered preparing and using the tris[(4-dimethylamino-2-pyridyl)methyl]amine (TPMA-NMe2) ligand in order to provide more active catalysts thereby implementing ligands with EDG for less active monomers such as vinyl acetate and N-vinyl pyrrolidone.
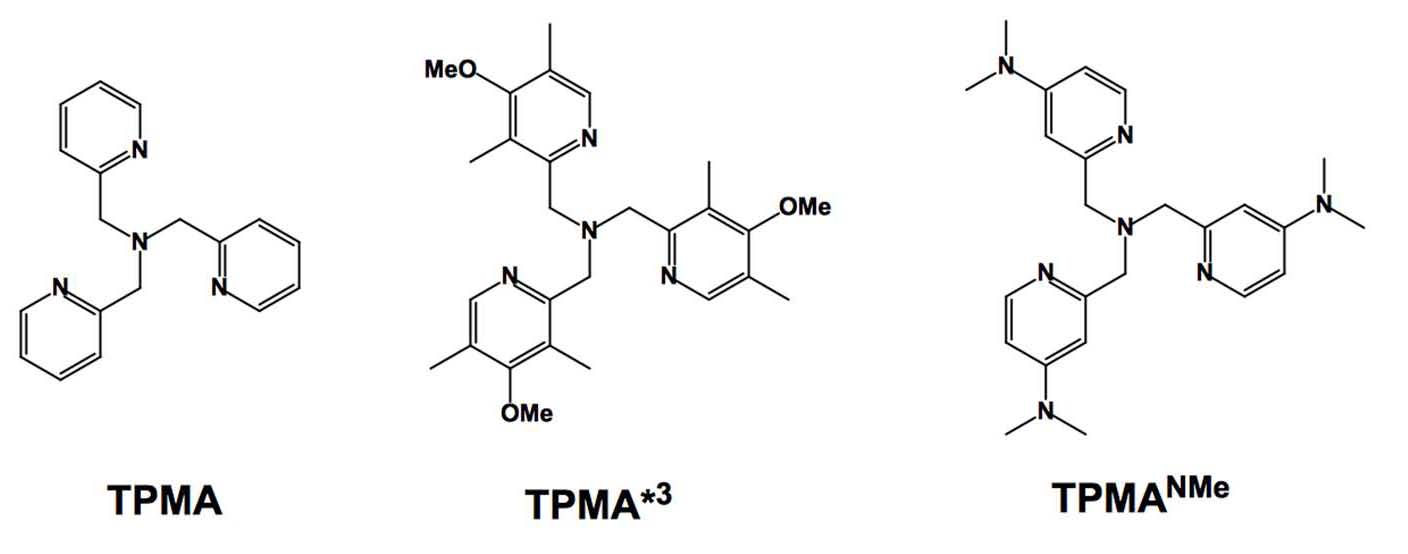
This new Cu based ATRP catalyst with TPMA-NMe2 ligands, in both relevant oxidation states, has been characterized in solution and in the solid state and utilized in ATRP systems with low catalyst loadings (down to 10 ppm) and was shown to exhibit unprecedented reactivity with alkyl halides. (20)
In order to assess the activity of the [Cu(TPMA-NMe)Br]+ catalyst, various forms of ATRP with activator regeneration were conducted. ICAR ATRP was conducted at catalyst loadings ranging from 100-10 ppm relative to monomer with 2,2′-azobis(2 methylpropionitrile) (AIBN) as radical initiator.(21) Linear semi-logarithmic plots vs. time were observed at all catalyst loadings. This is consistent with previously established ICAR kinetics with bimolecular termination for which the amount of catalyst does not change the rate of polymerization. (22) Over the course of 4 hours >90% conversion was achieved for all reactions with a linear increase in molecular weight with conversion, typical of a controlled polymerization. Although the rate of polymerization was unaffected by the initial amount of catalyst, molecular weight distributions gradually broadened upon decreasing catalyst concentration. This is attributed to a decreased rate of deactivation relative to propagation. Although the initial amount of catalyst does not change the rate of polymerization, at lower catalyst loadings, there is less deactivator complex to reversibly trap radicals. This results in more monomer additions per activation cycle and thus a broadening of molecular weight distributions.
Next, Ag0ATRP, a form of ARGET ATRP, was employed to further test the scope of the newly synthesized catalyst. Ag0ATRP uses silver wire to heterogeneously reduce L/CuII to L/CuI. The TPMA-NMe2 catalyst successfully polymerized MA via Ag0 ATRP with catalyst loadings down to 10 ppm. However, at 5 ppm, the polymerization was no longer living as observed by the decreasing molecular weights with conversion and high PDI values (>1.5). Current studies are currently being conducted to develop methods to decrease catalyst loadings to ppb levels.
Incorporating electron donating groups into phosphine based ligands provided a similar improvement in the activity of iron based catalysts for the polymerization of styrene. (18) In this paper the only EDG evaluated was a –OMe group and it was determined that FeIIIBr3 in the presence of tris(2,4,6-trimethoxyphenyl)-phosphine (TTMPP) provided faster ATRP of St than in the presence of tris(4-methoxyphenyl)phosphine and much faster than with triphenylphosphine under identical conditions. A ratio of ligand to iron of 2:1 provided a more active catalyst and the reaction progressed without the addition of any reducing agent indicating that the phosphines could directly reduce FeIII to FeII while also acting as ligands to form a complex with the transition metals and form efficient ATRP catalysts. However with a ratio on 2:1 with TTMPP ligand the molecular weight became higher than the theoretical value after 70% conversion, indicating some plausible contribution of termination by coupling. The reducing effect of the phosphine ligands and the possibility of coupling was confirmed by activation of PSt-Br macroinitiator with FeIIIBr3/TTMPP catalyst to form PSt with doubled molecular weight via coupling. ATRP of BA and MMA with the same catalysts containing the same phosphine ligands also provided well controlled reactions under proper conditions.
In conclusion, very active catalysts formed by incorporating EDGs into aromatic based ligands provide significantly better performance when combined with methods which require only ppm of catalyst and regenerate the active Cu(I)/ligand species during the polymerization, than with normal ATRP.
REFERENCES(1) Pintauer, T.; Matyjaszewski, K. Coordination Chemistry Reviews 2005, 249, 1155-1184.
(2) Tang, W.; Kwak, Y.; Braunecker, W.; Tsarevsky, N. V.; Coote, M. L.; Matyjaszewski, K. J. Am. Chem. Soc. 2008, 130, 10702-10713.
(3) Pintauer, T.; Matyjaszewski, K. Encycl. Radicals Chem., Biol. Mater. 2012, 4, 1851-1894.
(4) Pintauer, T.; Matyjaszewski, K. Top. Organomet. Chem. 2009, 26, 221-251.
(5) Kaur, A.; Ribelli, T. G.; Schroder, K.; Matyjaszewski, K.; Pintauer, T.: Inorg. Chem. 2015, 54 (4), 1474-1486.
(6) Matyjaszewski et al Macromolecules, 2016 49 pp 7749.
(7) Nanda, A. K.; Matyjaszewski, K. Macromolecules 2003, 36, 599-604.
(8) Nanda, A. K.; Matyjaszewski, K. Macromolecules 2003, 36, 1487-1493.
(9) Pintauer, T.; Reinoehl, U.; Feth, M.; Bertagnolli, H.; Matyjaszewski, K. European Journal of Inorganic Chemistry 2003, 2082-2094.
(10) Tang, H.; Arulsamy, N.; Radosz, M.; Shen, Y.; Tsarevsky, N. V.; Braunecker, W. A.; Tang, W.; Matyjaszewski, K. Journal of the American Chemical Society 2006, 128, 16277-16285.
(11) Pintauer, T.; Jasieczek, C. B.; Matyjaszewski, K. J. Mass Spectrom. 2000, 35, 1295-1299.
(12) Kickelbick, G.; Pintauer, T.; Matyjaszewski, K. New Journal of Chemistry 2002, 26, 462-468.
(13) Patten, T. E.; Xia, J.; Abernathy, T.; Matyjaszewski, K. Science (Washington, D. C.) 1996, 272, 866-868.
(14) Matyjaszewski, K.; Patten, T. E.; Xia, J. J. Am. Chem. Soc. 1997, 119, 674-680.
(15) Percec, V.; Barboiu, B.; Neumann, A.; Ronda, J. C.; Zhao, M. Macromolecules 1996, 29, 3665-3668.
(16) Xia, J.; Zhang, X.; Matyjaszewski, K. ACS Symp. Ser. 2000, 760, 207-223.
(17) Magenau, A. J. D.; Kwak, Y.; Schroder, K.; Matyjaszewski, K. ACS Macro Lett. 2012, 1, 508-512.
(18) Hansch, C.; Leo, A.; Taft, R. W. Chemical Reviews 1991, 91, 165-195.
(19) Schröder, K.; Mathers, R. T.; Buback, J.; Konkolewicz, D.; Magenau, A. J. D.; Matyjaszewski, K. ACS Macro Lett. 2012, 1, 1037−1040.
(20) Ribelli T. G.; Fantin, M.; Augustine K. F.; Matyjaszewski, K.; Daran, J.-C.; Poli, R.; J Am Chem Soc 2018, 140 (4), 1525-1534.
(21) D'Hooge, D. R.; Konkolewicz, D.; Reyniers, M.-F.; Marin, G. B.; Matyjaszewski, K.; Macromol. Theory Simul. 2012, 21 (1), 52–69.
(22) Krys, P.; Matyjaszewski, K.; European Polymer Journal 2017, 89, 482-523.
(23) Wang, Y.; Zhong, M.; Zhang, Y.; Magenau, A. J. D.; Matyjaszewski, K.; Macromolecules 2012, 45, 8929−8932.
(24) Wang, Y.; Soerensen, N.; Zhong, M.; Schroeder, H.; Buback, M.; Matyjaszewski, K.; Macromolecules 2013, 46 (3), 683-691.
(25) Wang, Y.; Kwak, Y.; Matyjaszewski, K. Macromolecules, 2012, 45 (15) 5911-5915.