Graft Copolymers with Complex Architecture
The accepted nomenclature is that one transitions from "graft" copolymers to "brush" copolymers when the density of the tethered side chains starts to affect the ability of the backbone of the copolymer to assume a random coil configuration.

AFM: M. Moeller & S. Sheiko (1)
Densely grafted copolymers
-
Linear macromolecular brushes
-
Star brush copolymers
-
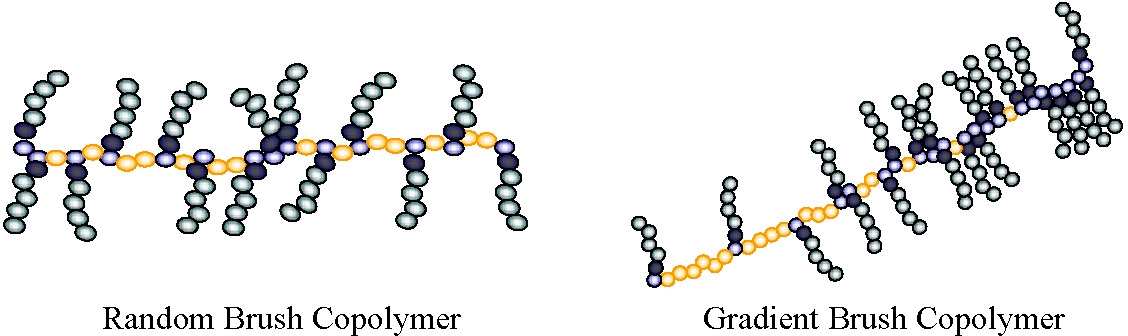
Gradient brush Copolymers
-
Brush block copolymers
-
Heterografted brush copolymers
-
Double grafted brush copolymers
-
Incorporation of site specific functionality
Densely grafted copolymers
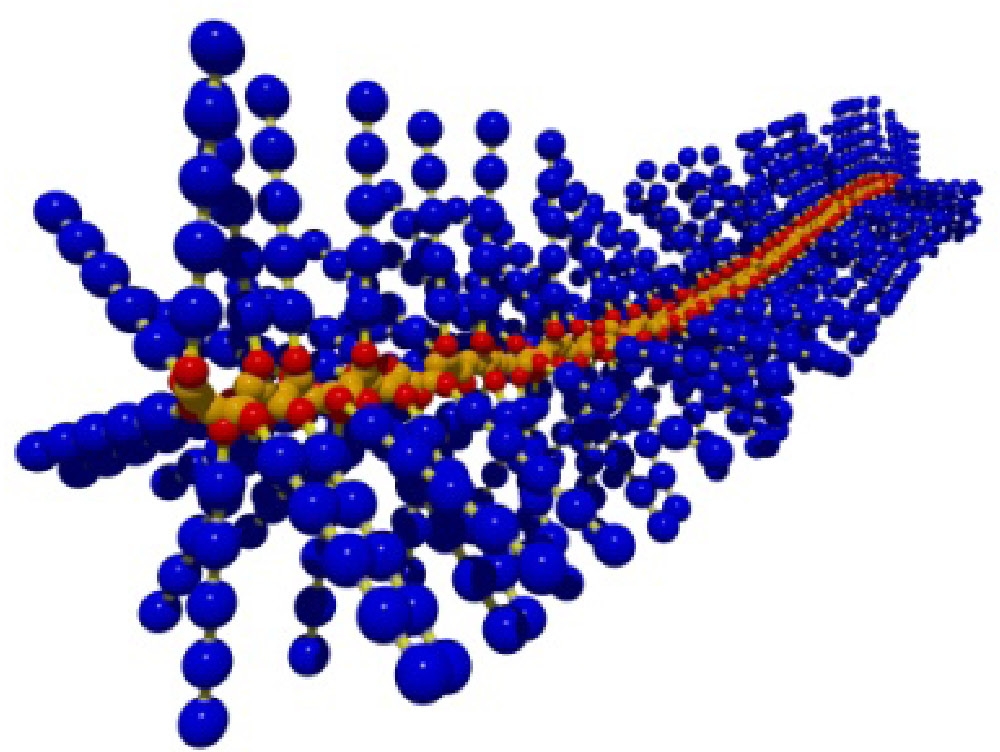
Linear brush-like molecular architectures are well-known in biology where they are responsible for various functions including mucociliary clearance of lung airways and mechanical performance of articular cartilage. As detailed below polymer chemistry is currently taking the first steps towards controlling all aspects of molecular architecture, understanding the distinctive properties of molecular brushes and how the physical properties of a macromolecule are affected by composition and topology.
One aspect of the group's research focuses on the synthesis of macromolecules with advanced intramolecular architectural control, particularly the synthesis and properties of macromolecular brushes, also called linear bottle-brush copolymers.
The synthesis of polymers with this type of complex molecular architecture by ATRP was initially an intellectual and synthetic challenge, perhaps subsequently stimulated by the visual images of individual macromolecules on adsorbing surfaces,(1) but has resulted in the preparation of materials with interesting physical properties.(2) Brush copolymers with block copolymer tethered chains provide environments for intramolecular chemistry, such as the formation of nanowires.(3,4)
The materials can be prepared by several techniques including "grafting through" and "grafting to" (5-9) but the following discussion focuses on "grafting from" well defined multifunctional linear and star macroinitiators(1) since this is the best approach for the synthesis of well defined densely grafted bottle-brush copolymers with pre-selectable topology, although the other approaches will be mentioned.
In order to form a molecule with a cylindrical shape, as shown in the majority of schematic images of brush macromolecules, the backbone should be much longer than the side chains.
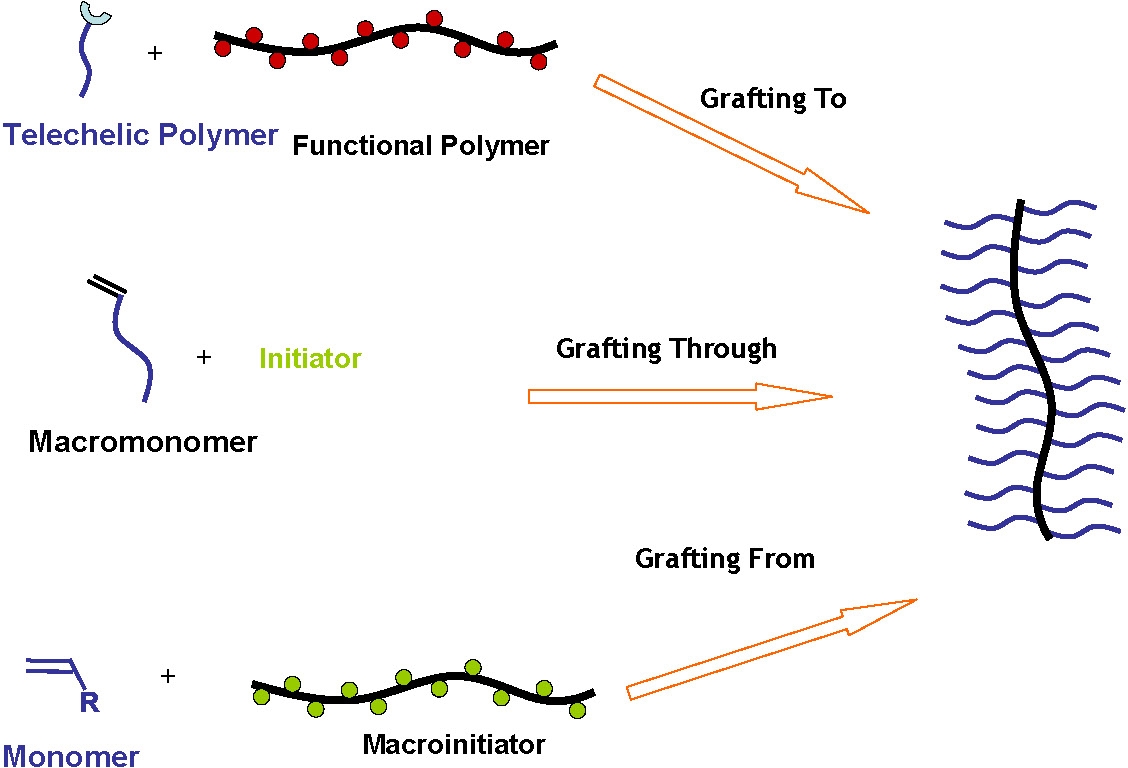
Grafting to reactions require the coupling of individual preformed tele-functional side chain precursors to a backbone polymer with distributed complementary functionality.
Grafting through consists of the polymerization of macromonomers.
Grafting from involves preparing the precursor to the backbone polymer with monomer units that contain functionalities ultimately capable of initiating polymerization of a second monomer.
Macromolecular brushes belong to the general class of graft copolymers. However, in this case the grafting density can be very high, at least in some segments of the copolymer. Indeed, polymers with one graft per backbone repeat unit have been prepared. This leads to an extremely crowded environment along the backbone which causes the macromolecules to adopt unusual conformations due to steric repulsion caused by the densely packed side chains which force the backbone from the normal Gaussian random coil conformation into a chain extended conformation with increased persistence length as side chain graft density increases.(10) The dynamic homogeneity of bottlebrush polymers with n-butyl acrylate side chains was discussed in a recent paper where it was determined that all short range correlations are dominated by the PBA side chains and long range correlations reflect the screening of the backbone by the tethered side chains(11) and confirmed that the equilibrium conformation results from the interplay between the repulsive forces due to steric congestion of the side chains close to the backbone and the entropic force of the backbone polymer. Two brush copolymers with side chain DP = 10 and 30 were examined by DSC, X-Ray scattering and rheology. The densely grafted side chains do stiffen the backbone but it retains some conformational freedom. The dynamics of the side chains and backbone are altered compared to the respective homopolymers, the backbone dynamics indicate they are plasticized by the side chains and the molecule becomes dynamically more homogeneous.
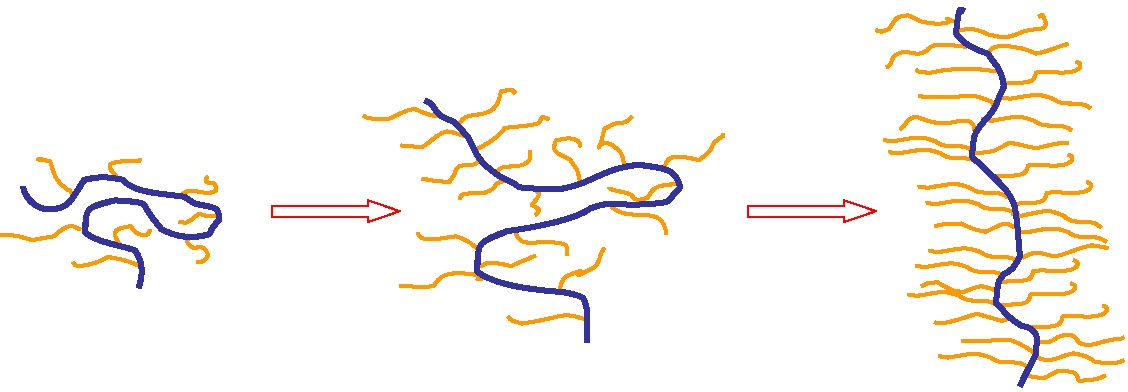
The congested structure of the brushes arises because of the confined mobility of the side chains.
A recent review(12) summarizes the characteristic physical properties of well-defined molecular brushes and the different strategies employed for their preparation (detailed below) with particular focus on synthesis via controlled radical polymerization techniques.
The environmentally benign activators generated by electron transfer for atom transfer radical polymerization (AGET ATRP) of oligo(ethylene glycol) monomethyl ether methacrylate (OEOMA) was investigated in homogeneous aqueous solution targeting DP = 1000, and in inverse miniemulsion targeting DP = 600, at 30 0C targeting biocompatible, brush-like, high MW, water-soluble polymers. The results indicated that AGET ATRP retains all of the benefits of normal ATRP and, additionally provides a facile route for the preparation of well-controlled high MW polymers because of the use of oxidatively stable catalyst precursors.(13)
Linear Macromolecular Brushes:
Synthesis of macromolecular brush copolymers takes advantage of all of the characteristics of CRP synthesis in that the molecular weight and composition of the backbone and attached side chains are independently controlled. Additionally, as shown for other polymers prepared by CRP, polymer brushes can be prepared as random graft copolymers, block graft copolymers,(14) gradient brush copolymers(15-16) and molecules with double grafted side chains (a brush of brushes).(17)
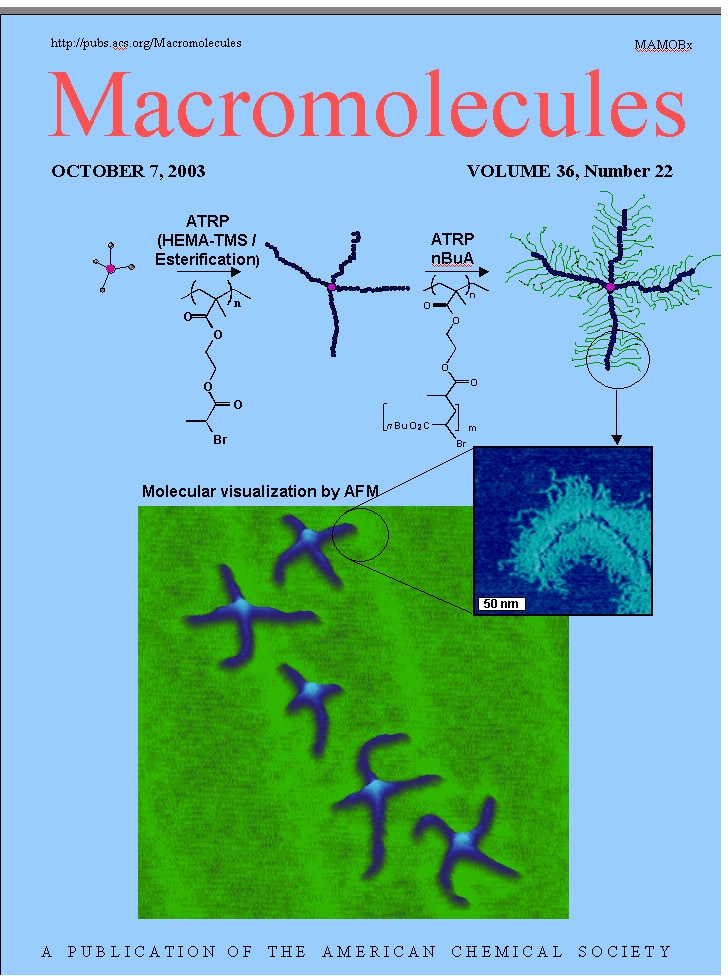
The first step in a typical preparation of a densely grafted bottle brush macromolecule is the preparation of a macroinitiator, or macroinitiator segment in a copolymer, with an initiating moiety on a high fraction of backbone monomer units. A macroinitiator for ATRP has generally been prepared by CRP of a monomer with a protected functional group, such as 2-(trimethylsilyloxy)ethyl methacrylate (HEMA-TMS), followed by cleavage of the TMS protective groups and esterification with 2-bromopropionyl bromide to yield poly(2-(2-bromopropionyloxy)ethyl methacrylate) (PBPEM).(7) This approach to brush copolymers is shown in the following schematic where the multifunctional macroinitiator is prepared and used to initiate ATRP of various monomers from each repeat unit by a grafting from mechanism.
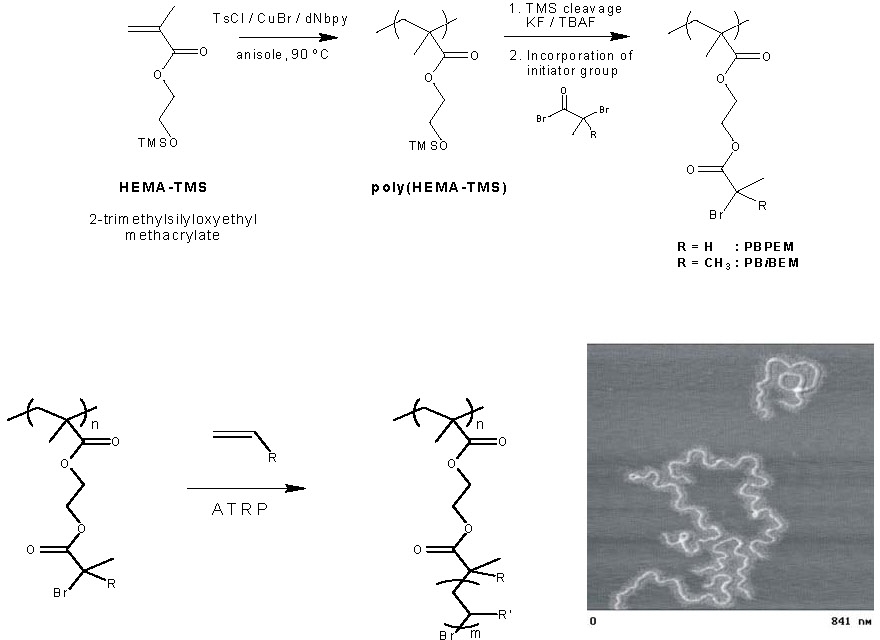
Macroinitiators with a backbone length of 50 to over 6,000 monomer units have been prepared by CRP processes and multiple graft chains, also via CRP, with 20 to over 400 monomer units have been prepared by grafting from reactions. Note that the molecular weight of a butyl acrylate based polymer with a backbone of 6,000 monomer units and 170 monomer units in each grafted side chain is well over 107. As seen in the image above, a brush macromolecule with a high DP of the backbone adopts a chain extended random coil conformation when a dilute solution of the brush molecule is deposited on an adsorbing surface despite the congested steric environment along the polymer backbone.
Generally the side chain density is controlled by controlling the distribution of initiating sites along the backbone by copolymerization of monomers with precursors of the initiating groups and non-initiating comonomers. The distribution of initiating sites can be further controlled by control over monomer feed and selection of monomers with favorable reactivity ratios of the initiating monomers and non-initiating comonomers. However, initiation efficiency also affects graft density. Grafting efficiency was generally assumed to be high because of the appearance of the final polymers. However by comparing the molecular weight of the cleaved side chains to their theoretical molecular weights, calculated assuming quantitative initiation, grafting efficiency was detected as a function of monomer conversion.(15-16)
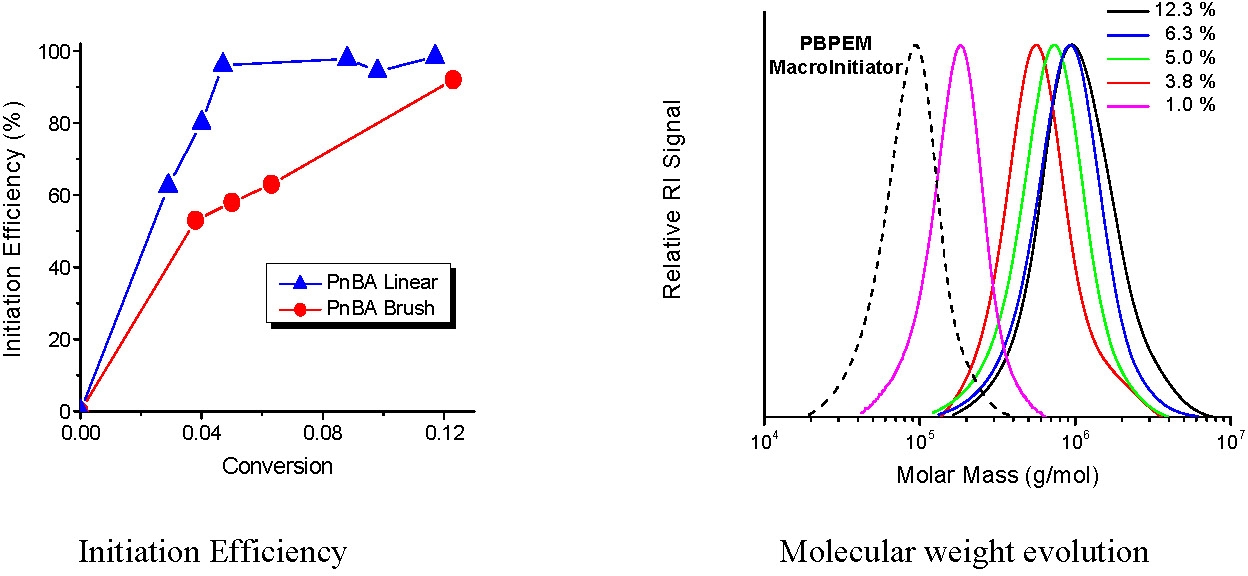
The results were compared to an analogous linear ATRP conducted under identical conditions, except that ethyl 2-bromopropionate was employed as the initiator. In the early stages of the polymerization initiation efficiency is lower than that observed from a low molecular weight mono-functional initiator but gradually increases to 87% at 12% monomer conversion. The difference was attributed to the congested environment, high local concentration of initiation sites, encountered in the case of grafting from a macroinitiator backbone, which leads to slower deactivation of growing radicals at low conversion. The initiation efficiency was enhanced by increasing the rate of deactivation of the growing species or by decreasing the rate of propagation(16) accomplished by either adding more Cu(II) or a lower concentration of the initiator to the reaction. A low concentration of active chain ends, [P*], suppresses intra- and inter-molecular termination and the activation / deactivation equilibrium which mitigates polymerization allowing all side chains to grow simultaneously.
Controlled copolymer synthesis and post-polymerization modification allows the molecular composition of the side chains to be tailored to the target application.
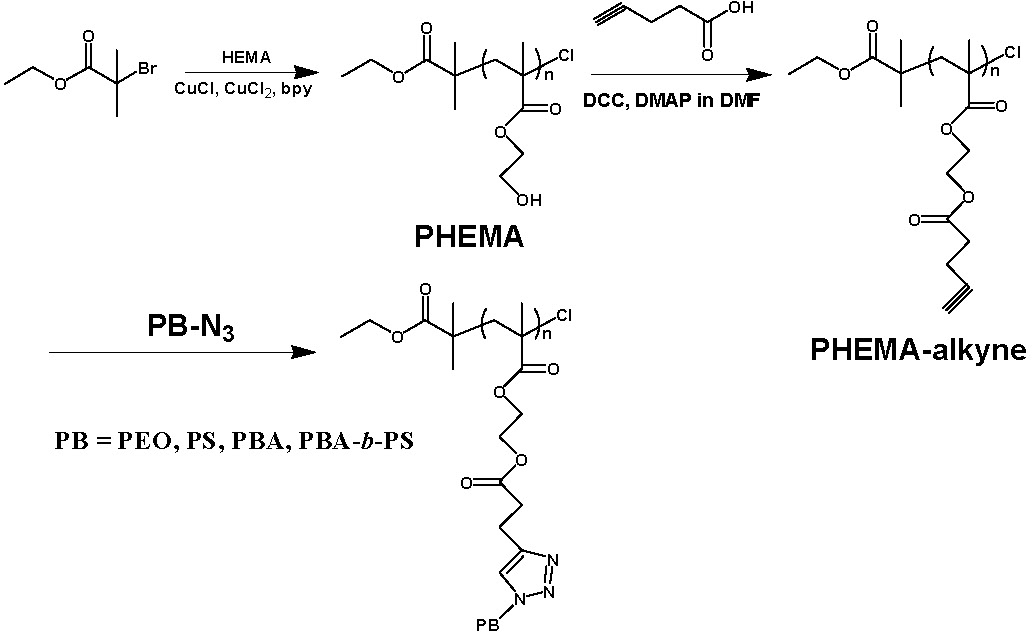
Recently,(5-6) an efficient grafting to approach has been successfully developed for the preparation of densely grafted brushes. The specific approach is not the only approach for grafting to but (perhaps) was selected because it operated using a copper catalyst complex. Linear poly(2-hydroxyethyl methacrylate) (PHEMA) polymers were synthesized first by ATRP. After esterification reactions between pentynoic acid and the hydroxyl side groups, polymeric backbones with alkynyl side groups on essentially every monomer unit (PHEMA-alkyne) were obtained. Five different kinds of azido-terminated polymeric side chains (SCs) with different chemical composition and molecular weight were used in the grafting to reactions, including poly(ethylene glycol)-N3 (PEO-N3), polystyrene-N3, poly(Bu acrylate)-N3, and poly(Bu acrylate)-b-polystyrene-N3. All click coupling reactions between alkyne-containing polymeric backbones (PHEMA-alkyne) and azido-terminated polymeric SCs were completed within 3 hours. The grafting density of the molecular brushes obtained was affected by several factors, including the molecular weight and the chemical structure of the linear polymers used in the grafting to reaction, as well as the initial molar ratio of linear chains to alkynyl groups. When linear polymers with "thinner" structure and lower molecular weight, e.g., PEO-N3 with Mn = 775 g/mol, were reacted with PHEMA-alkyne (DP = 210) at a high molar ratio of linear chains to alkynyl groups in the backbone, brush copolymers with the highest grafting density were obtained (efficiency of grafting = 88%). This result indicates that the average number of tethered side chains was ~ 186 per brush.
The AFM images shown on this page and in cited references generally show the brush copolymer deposited on a substrate that favors side chain adsorption. In this situation the molecule assumes a flat topology with chain extended side chains. This can present a strain on the backbone of the brush macromolecule as was demonstrated when inclusion of a weaker S-S bond allowed one to selectively break a specific covalent bond by adsorption-induced tension.(18)
Additional work(19) indicated that controlling the flow profile of a brush macromolecule fluid generates and delineates the concentration of mechanical forces enabling a hierarchical activation of all chemical bonds on different length scales from the macroscopic to the molecular scale. Bond tension is spontaneously generated within brushlike macromolecules as they spread on a solid substrate and the molecular architecture creates an uneven distribution of tension in the backbone covalent bonds, leading to spatially controlled bond scission. By controlling the flow rate and the gradient of the film pressure, one can sever the flowing macromolecules with high precision. Specific chemical bonds are activated within distinct macromolecules located in a defined area of a thin film. Furthermore, the flow-controlled loading rate enables quantitative analysis of the bond activation parameters. The intrinsic tension increases with grafting density, side chain length, and strength of adhesion to the substrate.(20) An extension of this study on the amplification of intrinsic tension upon specific bonds after adsorption of brush molecules onto a substrate was employed to study the effect of additional mechanical force on the kinetics of disulfide reduction in the presence of dithiothreitol. The cleavage reaction was investigated as a function of dithiothreitol concentration and backbone tension. The scission reaction of bottlebrush macromolecules containing a disulfide linker in the middle of the backbone was monitored through molecular imaging by AFM. The scission rate constant increased linearly with the concentration of dithiothreitol in the contacting solvent and exponentially with mechanical tension at the site of the disulfide bond. Furthermore, the rate constant at zero force was found to be significantly lower than the reduction rate constant in bulk solution, which suggests the surface of water is more acidic that bulk water and has acidic composition with a pH = 3.7. This work demonstrates the ability of branched macromolecules to accelerate chemical reactions at specific covalent bonds without applying an external force.
Star Brush Copolymers:
The use of a multifunctional initiator for the synthesis of the first macroinitiator backbone copolymer can lead to the preparation of bottle bush polymers displaying different topologies. For example, if a tetra-functional initiator is used to polymerize HEMA-TMS, the resulting polymer can be functionalized to yield a 4-arm macroinitiator. This product can, in turn, be used to prepare 4-arm star macromolecular brushes.(21) An AFM image of such a copolymer with poly(n-butyl acrylate) side chains is shown below.
Gradient Brush Copolymers:
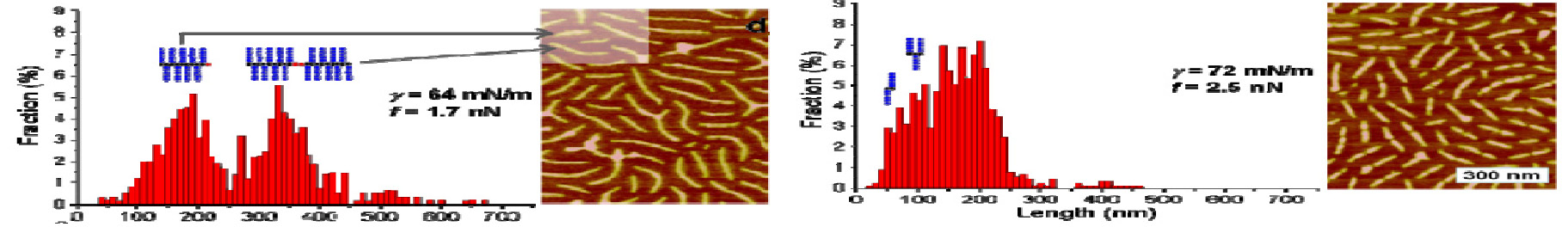
Combining gradient copolymerization with macroinitiator synthesis provides access to brushes with a controlled gradient of graft density along the back bone.(14-15) Gradient macroinitiators can be formed by spontaneous copolymerization of monomers with different reactivity ratios or by controlled addition of one monomer to a copolymerization of monomers with relatively similar reactivity ratios.(22) When a macroinitiator with a gradient of initiating sites along the backbone is used in a grafting from copolymerization, the resulting molecule has a higher density of side chains at one end of the backbone than it does at the other. AFM examination of gradient graft macromolecules after they are adsorbed on a mica substrate shows coexistence of two conformational phases within individual molecules. These observations were made by compressing mono-layers of the gradient brush macromolecules on the surface of water and then transferring a sample of this monolayer to a mica substrate for AFM studies. Upon compression, a rod-globule transition occurs at the end where the brush is densely grafted, leaving a molecule with a globular “head” and an extended “tail” a so-called “tadpole conformation.”(15)


A series of tadpole-shaped block-graft amphiphilic copolymers, i.e., block copolymers consisting of a cylindrical hydrophilic brush block and a coiled hydrophobic block were synthesized using "grafting-through" ATRP.(23) A tadpole-shaped block-graft copolymer from polystyrene bromide and a methacryloyl-terminated poly(tert-Bu acrylate) was prepared first then, hydrolysis of the poly(tert-Bu acrylate) side chains to polyacrylic acid side chains provided tadpole-shaped block-graft amphiphilic copolymers, which formed pH responsive micelles in water, the latter being confirmed by dynamic light scattering and AFM.
Brush Block Copolymers:
Topologically different materials can be described as brush block copolymers including:
a) a brush macromolecule with block copolymer grafted side chains and
b) a “standard block copolymer” where one or more segments along a polymer backbone are brush copolymers of different composition.
a) Block copolymer side chains: The use of a macromolecular brush as a macroinitiator to polymerize a second monomer allows the synthesis of densely grafted copolymers with block copolymer side chains.(10)
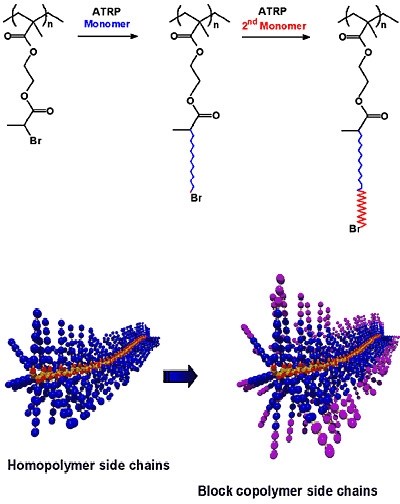
The resulting nanostructured macromolecules can form soft/hard core/shell systems (e.g. poly(n-butyl acrylate) / polystyrene) and potentially also macromolecular channels, stable worm-like micelles or inverse micelles, and other complex architectures. These materials have served as templates for the preparation of gold nanowires.(3)
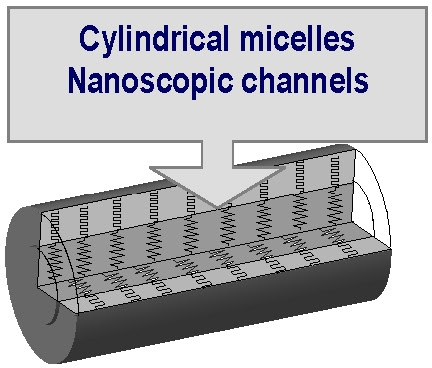
The molecular shape and the intramolecular phase behavior of collapsed single molecular brushes with homo- and block copolymer side chains can be studied by AFM and the conformation changes from rod like to globular upon decrease of the surface energy of the substrate. The conformational changes result from partial desorption of poly(Bu acrylate) side chains as the surface pressure drops from 23.7 to 3.1 mN/m and the energy of interaction between the side chains and the substrate decreases from 89.7 mJ/m2 to 69.1 mJ/m2. In the images below, brush copolymers with poly(n-butyl acrylate-block-styrene) side chains cast onto a mica surface are shown. The polystyrene segments desorb from the surface and form regular nano-domains atop the extended copolymer backbone.(15)
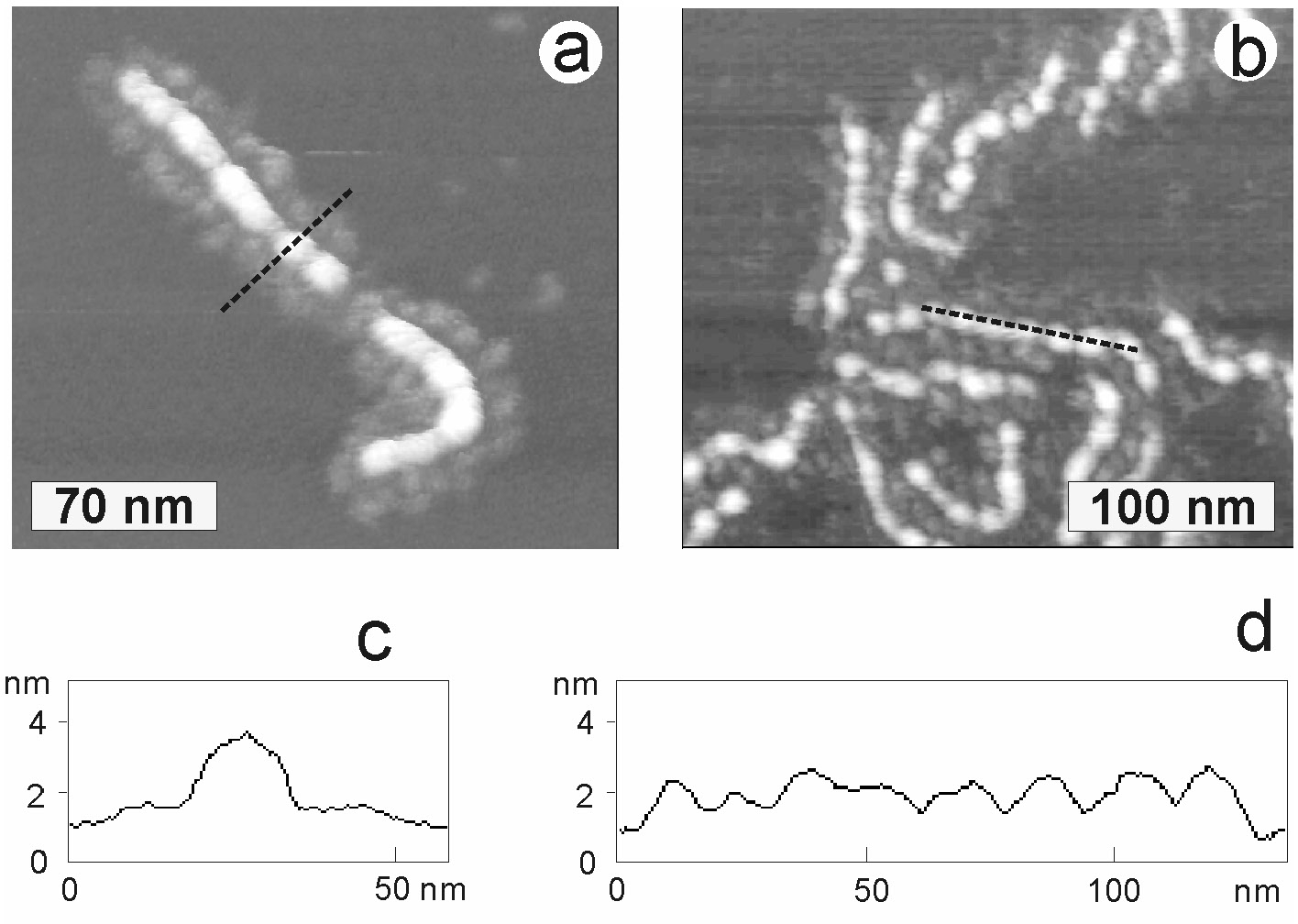
A dual mechanism polymerization (ATRP/RAFT) has also been applied to the preparation of brush copolymers with block copolymer side chains.(24) The “standard” backbone hydroxy functional macromolecule, poly(2-hydroxyethyl methacrylate) was converted into a macro chain transfer agent poly(2-((2-ethylxanthatepropanoyl)oxy)ethyl methacrylate) (PXPEM) by attaching xanthate chain transfer agents onto each monomeric unit using a DCC coupling reaction. Subsequently, a RAFT polymerization procedure was used to synthesize molecular bottlebrushes with poly(N-vinylpyrrolidone) side chains with controlled MW and low polydispersity by grafting from the PXPEM backbone. A low concentration of free radical initiator was employed at low temperature to minimize radical concentration in the system, which is an essential requirement that minimizes radical termination reactions (both intramolecular and intermolecular) and also to decrease the amount of radical initiated new chains. The side chains, DP ~40, were then chain extended with vinyl acetate under yielding well defined bottlebrush macromolecule with PNVP-b-PVOAc side chains when conversion of vinyl acetate was held to low levels, ~10%. The comb-like shape of the chain extended bottlebrushes was confirmed by AFM where it was noted that relatively large amounts of linear polymer chains were formed due to transfer reactions and new chains generated by AIBN during the side chain growth. These free polymer chains caused void formation in the AFM-LB film images.

Side chain DP = 130, 40% linear polymer Side chain DP 40, 9% linear polymer
b) Block copolymer backbone:
The second approach to prepare a material that can also be described as a block brush copolymer, is the preparation of a macroinitiator precursor followed by chain extension with another monomer. This leads to “standard” block copolymer architecture along the backbone of the brush. The second block can be used to form a phase separable segment to physically link the bottle brush copolymer macromolecules, e.g. in the following schematic and AFM image show association behavior between two terminal ODMA units driven by crystallization.
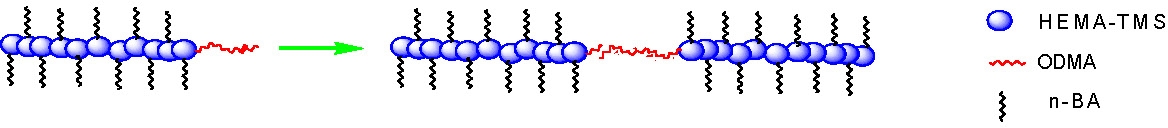
AFM image of a brush copolymer with linear block copolymer architecture along the backbone. The first block has pendant n-BA chains while the second block is composed of ODMA monomer units and causes end-to-end phase separation and physical linking of two or more brush macromolecules as the polymer is cast from dilute solution.

A star block copolymer would be expected to form a network.
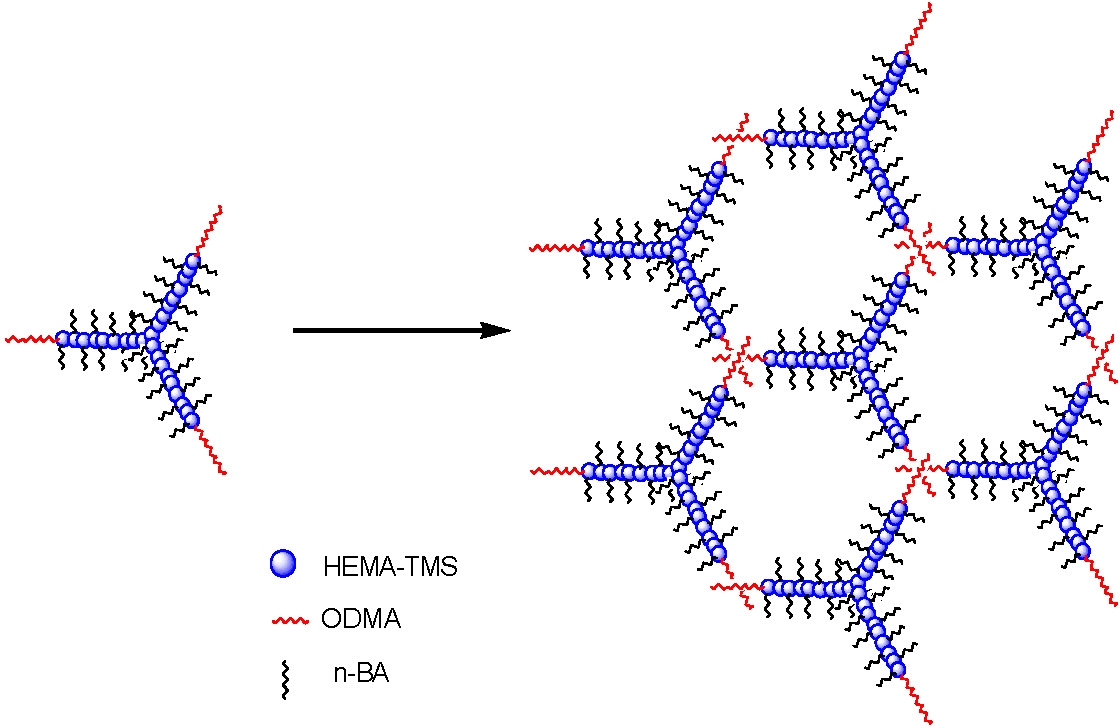
Indeed, in reference 19, it was reported that covalently crosslinked phase separated brush copolymers behaved as a thermoplastic SuperSoft elastomer. (See page 11)
Alternatively, the second block can also have brush architecture.
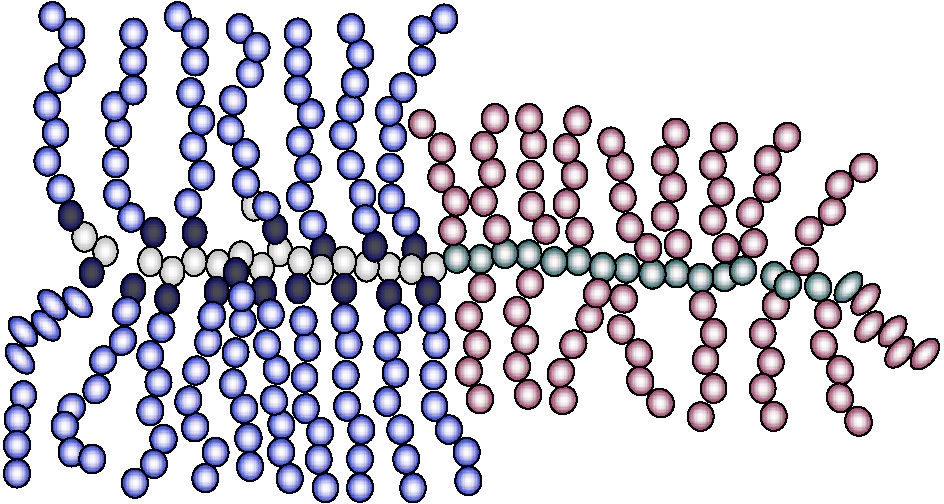
Block brush-copolymers can also be prepared by forming the backbone block copolymer with one segment of the block copolymer possessing functionality capable of initiating polymerization by a mechanism other than the CRP or by using a different CRP process. One example is a amphiphylic hetero-grafted block brush macromolecules with a crystalline poly(ε-caprolactone) (PCL) brush-like block and an amorphous poly(Bu acrylate) brush as the second block.(26) The block brush copolymer was synthesized entirely by the "grafting from" approach using a combination of ATRP and ring-opening polymerization (ROP). A well-defined poly(2-hydroxyethyl methacrylate) (PHEMA) was synthesized by ATRP and then the PHEMA was used as a macroinitiator for chain extension with 2-(trimethylsilyloxy)ethyl methacrylate (HEMATMS) via ATRP to provide PHEMA-b-PHEMATMS block copolymer. The functional groups in each block were transformed into initiators for ATRP and ROP. The PCL was grafted from the -PHEMA segment of the dual functional macroinitiator by ROP in the presence of tin(II) 2-ethylhexanoate catalyst then PBA chains were grafted from the brush macroinitiators by ATRP. AFM images show that because of their amphiphilic structure, the hetero-grafted diblock brushes demonstrated association into flower-like or dumbbell-like structures due to the association/crystallization of the PCL blocks.

Recently Rzayev (27) prepared polystyrene-b-polylactide bottlebrush block copolymers by a combination of three controlled polymerization techniques in order to study their self assembly into large domain ordered nanostructures. The block copolymer backbone for a dual grafting from polymerization was prepared by RAFT polymerization of solketal methacrylate (SM) followed by polymerization of 2-(bromoisobutyryl)ethyl methacrylate (BIEM). Polystyrene branches were grafted by ATRP from the poly(BIEM) block, and PLA branches were “grafted from” the poly(SM) block after the removal of ketal groups.
Earlier work by Ishizu had taken a different approach to preparing a similar structure by sequential copolymerization of a backbone precursor initiator monomer with a macromonomer.(17) This would lead to a block brush copolymer that was prepared by a combination of “grafting from” and “grafting through.”
Heterografted brush copolymers:
Another interesting brush topology is demonstrated by synthesis of materials, called hetero-grafted brush copolymers. This type of brush copolymer with grafts of different composition along the backbone was initially prepared by a random grafting through copolymerization of a macromonomer with a low molecular weight comonomer that was a precursor for a CRP initiator.(28-29)
The protecting groups on the precursor monomer units were derivatized to yield moieties capable of initiating ATRP, followed by a grafting from polymerization of a second monomer from the distributed initiating groups along the brush copolymer backbone.
A brush copolymer with poly(ethylene oxide) grafts and poly(butyl acrylate) grafts prepared as described above displays interesting behavior. The materials undergo a reversible collapse in the presence of ethanol or water vapors which results in a change of the surface properties of mica, favoring either adsorption or desorption of one of the graft polymer segments.(30) When extended, strongly adsorbed poly(butyl acrylate) brush molecules are exposed to ethanol vapor, the macromolecules swell and contract to form compact globules. Exchanging the ethanol vapor with a humid atmosphere caused the molecules to extend again to a wormlike two-dimensional conformation.
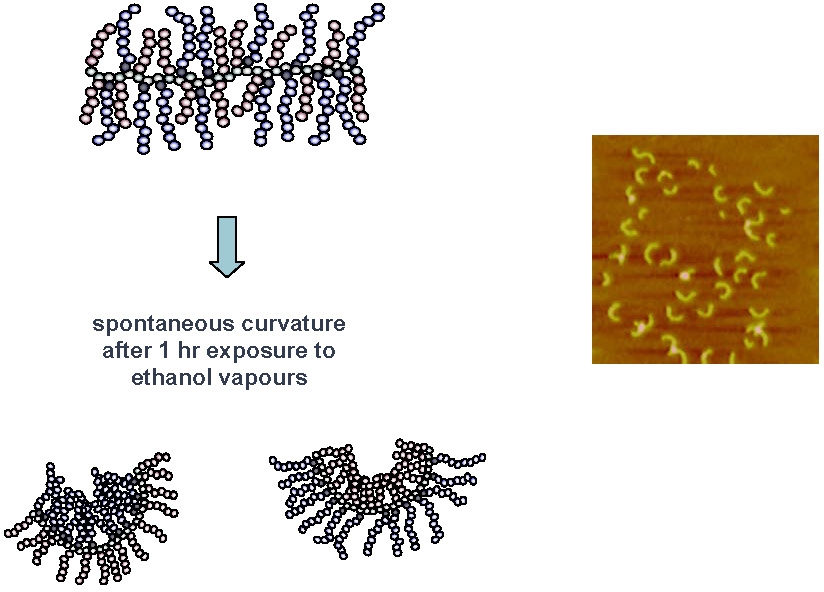
Polymer brush molecules with responsive units in the graft chains can be prepared by copolymerization.
Double Grafted Brush Copolymers:
Another combination of grafting from and grafting through can lead to the preparation of a type of copolymer named "double grafted" brush copolymers. This topology arises when macromonomers are polymerized by grafting from a linear macroinitiator.
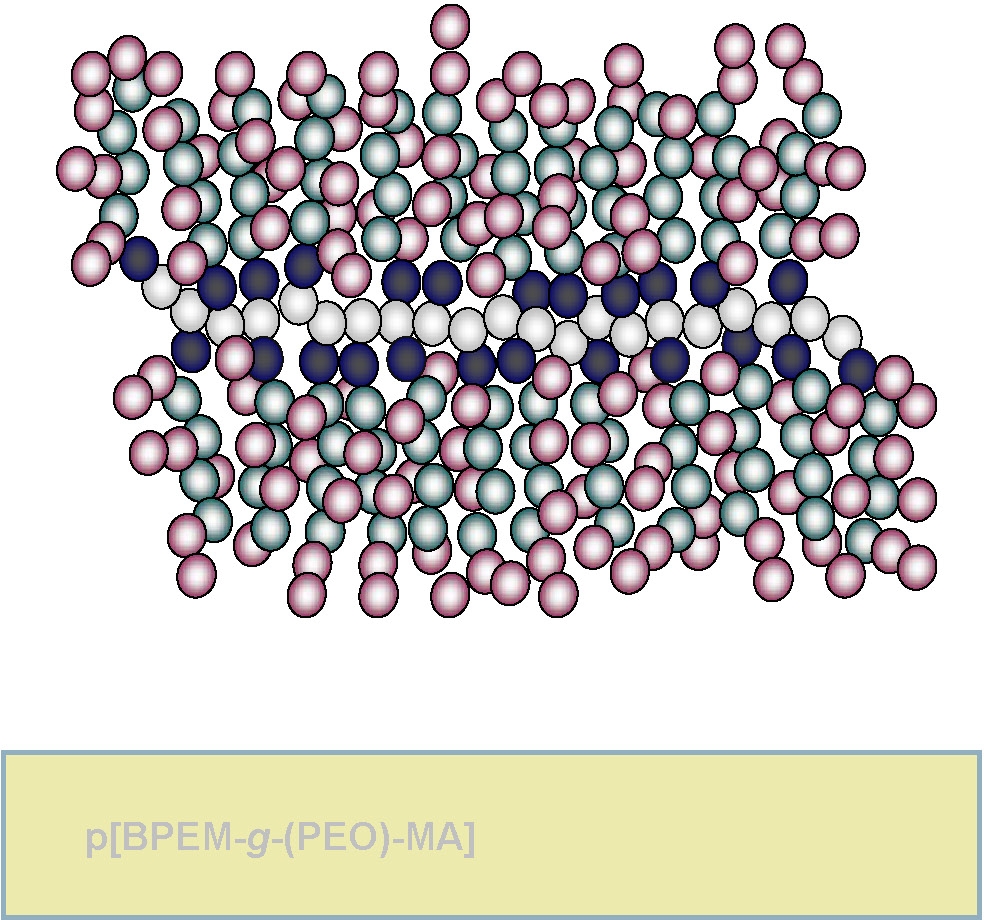
In many ways this can be considered a brush copolymer where each graft is itself a brush copolymer.(2)
Incorporation of site-specific functionality:
Site specific functionality is illustrated by formation of molecules with preselected functionality in a specific bond within the copolymer, functionality at the molecular level, and intramolecular functionality the molecular level, and interactive intermolecular functionality in bulk materials.
Functionality at the single bond level was exemplified by introduction of a disulfide bond in the middle of the bottlebrush backbone which was later cleaved by adsorption induced stress.(31) While covalent carbon-carbon bonds are harder to break the incorporation of a disulfide bond close to the center of a brush macromolecule illustrated that simple adsorption of brush-like macromolecules with long side chains on a substrate can induce not only conformational deformations described above, but also spontaneous rupture of covalent bonds in the backbone. This behavior was attributed to the fact that the attractive interaction between the side chains and the substrate is maximized by the spreading of the side chains, which in turn induces tension along the polymer backbone. Provided the side-chain densities and substrate interaction are sufficiently high, the tension generated was strong enough to rupture covalent carbon-carbon bonds. We expect similar adsorption-induced backbone scission to occur for all macromolecules with highly branched architectures, such as brushes and dendrimers. This behavior needs to be considered when designing surface-targeted macromolecules of this type-either to avoid undesired degradation, or to ensure rupture at predetermined intra-macromolecular sites.
A readily visible incorporated site specific functionality was attained by copolymerization of fluorescein O-methacrylate with Bu acrylate in a "grafting from" reaction with a multifunctional linear macroinitiator to form pH responsive fluorescent bottlebrushes.(32) The brush-like structure of the synthesized macromolecule was confirmed by AFM while NMR spectroscopy showed that the fluorescent units were successfully incorporated into the polymer. The synthesized bottlebrushes displayed highly fluorescent properties under basic conditions, yet showed no fluorescence under neutral or acidic conditions. The fluorescence could be turned on and off by changing the pH of the solution.
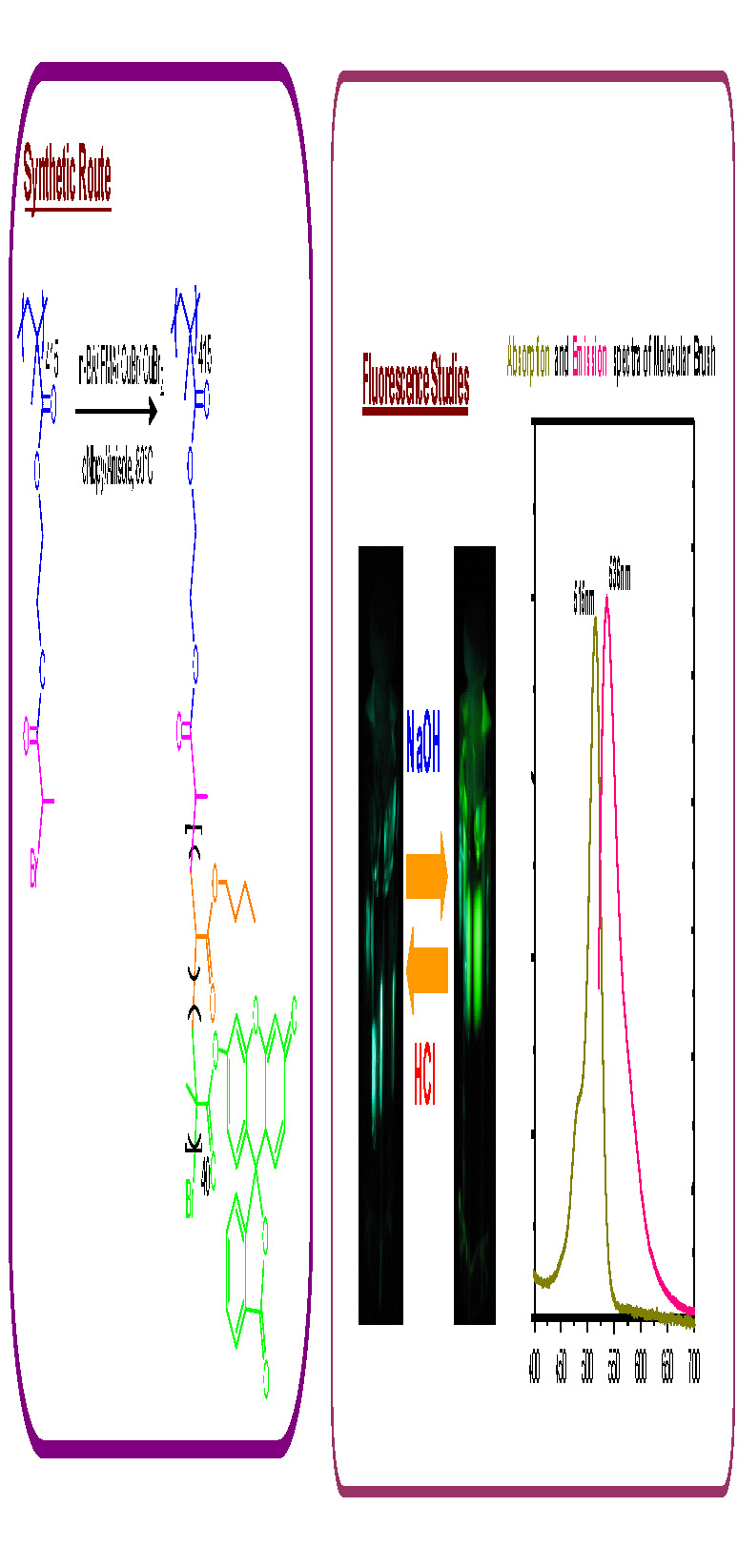
These bottlebrush molecules could have potential applications in molecular imaging.
REFERENCES
(1) Beers, K. L.; Gaynor, S. G.; Matyjaszewski, K.; Sheiko, S. S.; Moeller, M. Macromolecules 1998, 31, 9413-9415.(2) Neugebauer, D.; Zhang, Y.; Pakula, T.; Sheiko, S. S.; Matyjaszewski, K. Macromolecules 2003, 36, 6746-6755.
(3) Djalali, R.; Li, S.-Y.; Schmidt, M. Macromolecules 2002, 35, 4282-4288.
(4) Zhang, M.; Estournes, C.; Bietsch, W.; Mueller, A. H. E. Advanced Functional Materials 2004, 14, 871-882.
(5) Sumerlin, B. S.; Tsarevsky, N. V.; Louche, G.; Lee, R. Y.; Matyjaszewski, K. Macromolecules 2005, 38, 7540-7545.
(6) Gao, H.; Matyjaszewski, K. Journal of the American Chemical Society 2007, 129, 6633-6639.
(7) Boerner, H. G.; Matyjaszewski, K. Macromolecular Symposia 2002, 177, 1-15.
(8) Tsarevsky, N. V.; Bencherif, S. A.; Matyjaszewski, K. Macromolecules 2007, 40, 4439-4445.
(9) Van Camp, W.; Germonpre, V.; Mespouille, L.; Dubois, P.; Goethals, E. J.; Du Prez, F. E. Reactive & Functional Polymers 2007, 67, 1168-1180.
(10) Rathgeber, S.; Pakula, T.; Wilk, A.; Matyjaszewski, K.; Beers, K. L. Journal of Chemical Physics 2005, 122, 124904/124901-124904/124913.
(11) Grigoriadis, C.; Nese, A.; Matyjaszewski, K.; Pakula, T.; Butt, H.-J.; Floudas, G. Macromol. Chem. Phys. 2012, 213, 1311–1320,.
(12) Sheiko, S. S.; Sumerlin, B. S.; Matyjaszewski, K. Prog. Polym. Sci. 2008, 33, 759-785.
(13) Oh, J. K.; Perineau, F.; Charleux, B.; Matyjaszewski, K. J. Polym. Sci., Part A: Polym. Chem. 2009, 47, 1771-1781.
(14) Boerner, H. G.; Beers, K.; Matyjaszewski, K.; Sheiko, S. S.; Moeller, M. Macromolecules 2001, 34, 4375-4383.
(15) Boerner, H. G.; Duran, D.; Matyjaszewski, K.; da Silva, M.; Sheiko, S. S. Macromolecules 2002, 35, 3387-3394.
(16) Lord, S. J.; Sheiko, S. S.; LaRue, I.; Lee, H.-I.; Matyjaszewski, K. Macromolecules 2004, 37, 4235-4240.
(17) Ishizu, K.; Satoh, J.; Sogabe, A. Journal of Colloid and Interface Science 2004, 274, 472-479.
(18) Park, I.; Sheiko, S. S.; Nese, A.; Matyjaszewski, K. Macromolecules 2009, 42, 1805-1807.
(19) Park, I.; Shirvanyants, D.; Nese, A.; Matyjaszewski, K.; Rubinstein, M.; Sheiko, S. S. J. Am. Chem. Soc. 2010, 132, 12487-12491.
(20) Li, Y.-C.; Nese, A.; Lebedeva, N. V.; Davis, T.; Matyjaszewski, K.; Sheiko, S. S. J. Am. Chem. Soc. 2011, 133, 17479–17484.
(21) Boyce, J. R.; Shirvanyants, D.; Sheiko, S. S.; Ivanov, D. A.; Qin, S.; Boerner, H.; Matyjaszewski, K. Langmuir 2004, 20, 6005-6011.
(22) Kajiwara, A.; Matyjaszewski, K.; Kamachi, M. ACS Symp. Ser. 2000, 768, 68-81.
(23) Ohno, S.; Nese, A.; Cusick, B.; Kowalski, T.; Matyjaszewski, K. Vysokomol. Soedin., Ser. A Ser. B 2009, 51, 1947-1954.
(24) Nese, A.; Li, Y.; Averick, S.; Kwak, Y.; Konkolewicz, D.; Sheiko, S. S.; Matyjaszewski, K. ACS Macro Lett. 2012, 1, 227-231.
(25) Pakula, T.; Zhang, Y.; Matyjaszewski, K.; Lee, H.-i.; Boerner, H.; Qin, S.; Berry, G. C. Polymer 2006, 47, 7198-7206.
(26) Lee, H.-i.; Matyjaszewski, K.; Yu-Su, S.; Sheiko, S. S. Macromolecules 2008, 41, 6073-6080.
(27) Rzayev, J. Macromolecules 2009, 42, 2135-2141.
(28) Neugebauer, D.; Zhang, Y.; Pakula, T.; Matyjaszewski, K. Polymer 2003, 44, 6863-6871.
(29) Neugebauer, D.; Theis, M.; Pakula, T.; Wegner, G.; Matyjaszewski, K. Macromolecules 2006, 39, 584-593.
(30) Gallyamov, M. O.; Tartsch, B.; Khokhlov, A. R.; Sheiko, S. S.; Boerner, H. G.; Matyjaszewski, K.; Moeller, M. Chemistry--A European Journal 2004, 10, 4599-4605.
(31) Sheiko, S. S.; Sun, F. C.; Randall, A.; Shirvanyants, D.; Rubinstein, M.; Lee, H.-i.; Matyjaszewski, K. Nature (London, United Kingdom) 2006, 440, 191-194.
(32) Nese, A.; Lebedeva, N. V.; Sherwood, G.; Averick, S.; Li, Y.; Gao, H.; Peteanu, L.; Sheiko, S. S.; Matyjaszewski, K. Macromolecules 2011, 44, 5905–5910.