Development of ATRP
Historically the Matyjaszewski group has focused on the development and use of copper based catalysts (1-4) for Atom Transfer Radical Polymerizations (ATRP) conducted with a halogen as the transferable atom. This work forms the foundation for most of the observations/discussions/conclusions on the other pages of this web site. Other transition metals have been examined, and continue to be examined both within the group and by many other researchers throughout the world, (1,5-12) and other transferable atoms or groups have also been explored. (13-19)
Mechanistic studies provide the fundamental foundation required to develop a comprehension of the critical parameters for an ATRP. They provided the knowledge required in order to develop a more environmentally benign ATRP procedure, and remain crucial to any future developments in ATRP, since they provide the kinetic data that provides the underpinnings for chemical engineers to scale up the procedures to viable industrial scale production of specialty materials.
Correlating reaction parameters, including measurement of the rate constants for initiation, activation, deactivation, and hence overall reaction rate; (20) and evolution of molecular weight and molecular weight distribution with selected initiator, monomer, transition metal, ligand and resulting catalyst structure, solvent composition, and temperature should ultimately lead to the development of more active catalysts that can be specifically selected for optimum controlled synthesis of targeted materials in both homogeneous and biphasic media under industrially scalable conditions. (21,22)
This means that one catalyst does not work for every monomer in every solvent or in every dispersed polymerization medium. Selection of an appropriate catalyst system, reaction medium, and reaction conditions are required. (23,24) For instance if conditions are defined employing a lower activity catalyst complex, such as a copper/N-(n-alkyl)-2-pyridylmethanimine, (25) they will not provide a controlled polymerization if they are applied to a polymerization with a catalyst complex that is a million times more active, such as a complex formed with Me6TREN. (26) This difference in level of control over the final PDI and differences in the final color of the added catalyst complex when two reactions with different catalysts are conducted under the same reaction conditions, which actually requires holding the reaction in the presence of the higher activity catalyst at high temperature and high conversion of monomers for many hours. This results in some side reactions. It does not mean a different mechanism is operating only that process conditions have to take into account the reaction parameters.
More active catalysts are less oxidatively stable and reactions should be conducted at lower temperature and lower catalyst levels in order to control the reaction. See "How to Conduct an ATRP" for some suggested starting points.
The general scheme depicting the mechanism of ATRP is shown below.
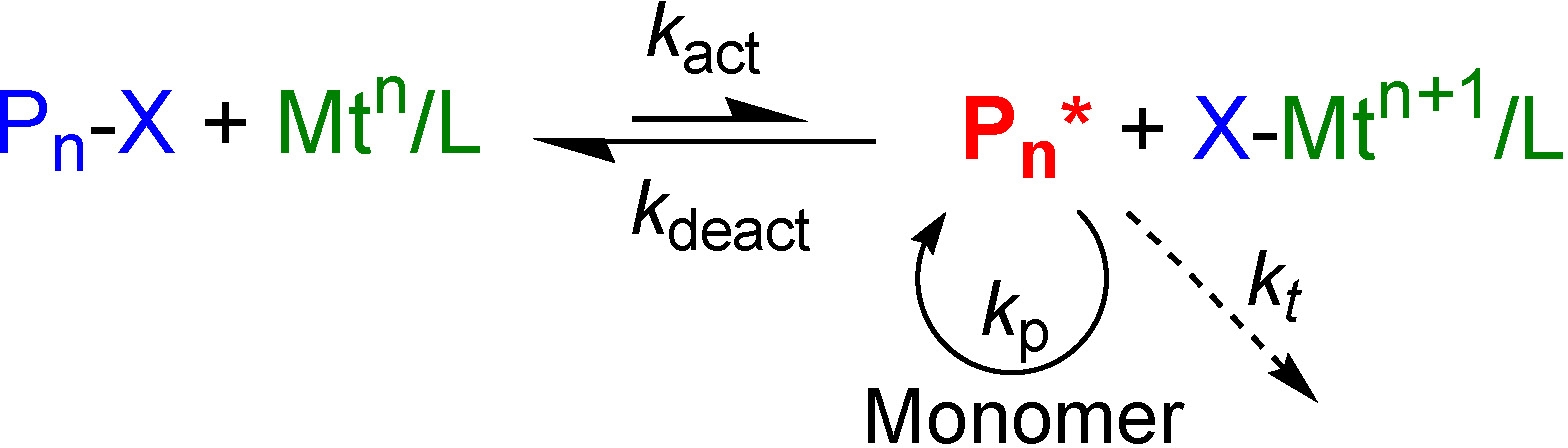
Most ATRP reactions require the addition, or in situ formation, of four essential components for an ATRP reaction: (27,28)
- a molecule, which we have called a (macro)initiator Pn-X, with at least one transferable atom or group, frequently a halogen where X = Cl or Br;
- a transition metal compound, Mtm, that can undergo a one electron redox reaction,
- a ligand, L, that forms a complex with the transition metal (compound) to modify catalyst solubility and catalyst activity, and
- one or more radically (co)polymerizable monomers.
Other language can, and has been used to describe this process. Indeed many authors have intermittently used other names/abbreviations for similar reactions utilizing the same components including: Living (or controlled) Radical Polymerization Mediated by Transition Metal complexes (LRP-MTM), Transition Metal-Mediated Living Radical Polymerization (TMM-LRP), Metal-Catalyzed Living Radical Polymerization (MC-LRP), Copper Mediated Living Radical Polymerization (CM-LRP), Single-Electron Transfer Living Radical Polymerization (SET-LRP) and recently, Outer Sphere Electron Transfer Metal-Catalyzed Polymerization (OSET-MCP). (8,12,29-35)
Rather than looking at prior art to provide a historically relevant name authors created many of these abbreviations to clarify their proposed mechanism and differentiate their work from similar efforts by others in the field. This may have created confusion as to the fundamental similarity, indeed identical nature, of the reactions being discussed. The basic principles underlying all controlled polymerization processes using the direct addition or in situ formation of these four reagents are the same. (36)
In all of the above listed variations in nomenclature the disclosed process involves polymerization of radically (co)polymerizable monomers by a procedure requiring formation of a dynamic equilibrium requiring Activation of a dormant species (reaction of a alkyl (pseudo)halide initiator with a lower oxidation state transition metal complex comprising a ligand) and Deactivation of the growing polymer chain end (reaction of the active propagating radical with transition metal complex in higher oxidation state comprising a radically transferable atom or group). All utilize transfer of a radically transferable atom or group from a molecule most call an "initiator" to a lower oxidation state transition metal complex in a homolytic bond cleavage reaction to form an active propagating species that can incorporate one or more monomers prior to being deactivated by reverse transfer of the atom or group to reform the dormant species or new initiator species. The basic principles underlying all processes using the direct addition or in situ formation of these four components are the same resulting in the same mechanism irrespective of the name given to the reaction.
On this web page we call it ATRP.
The existence of a radical, (Pn*), here we are using IUPAC nomenclature#, has been proposed in copper-mediated ATRP and is based on several experimental observations, (37) which are discussed below in greater detail.
# (http://www.iupac.org/goldbook/R05066.pdf),One or more of these functions can be combined in a single molecule, e.g. an initiator and monomer, (which directly forms a (hyper)branched structure when (co)polymerized (38)) or the role of solvent and (pseudo)ligand. (39,40)
Mechanistically, ATRP is based on an inner sphere electron transfer process, (41) which involves a reversible homolytic (pseudo)halogen transfer between a dormant species, an added initiator or the dormant propagating chain end, (Pn-X) and a transition metal complex in the lower oxidation state (Mtm/Ln) via a halogen-bridged transition state resulting in the formation of propagating radicals (Pn*) (42) and the metal complex in the higher oxidation state with a coordinated halide ligand (e.g. X-Mtm+1/Ln).
The active radicals form at a rate constant of activation (kact), subsequently propagate with a rate constant (kp) and reversibly deactivate (kdeact), but also terminate (kt). The rate of polymerization of a given monomer (M) depends on the radical concentration [Pn*] where radical concentration is further determined by the equilibrium constant (KATRP) for the ATRP. Targeted values of KATRP should be low (10-9~10-4) in order to maintain a low concentration of radicals in the polymerization medium and minimize termination reactions. The apparent KATRP (KATRPapp = KATRP/[CuII]) can be estimated from the kinetic plot of ln([M]0/[M]) vs. time for an ATRP experiment; (43) however, precise values for KATRP are difficult to measure during an actual polymerization reaction because CuII concentrations continuously change due to the persistent radical effect (PRE) (44) and the equilibrium is strongly shifted towards the dormant species (kact << kdeact). Recently equilibrium constants for copper-based ATRP were determined for a wide range of ligands and initiators in acetonitrile at 22 0C. The ATRP equilibrium constants obtained varied over 7 orders of magnitude and strongly depended on the structure of the ligand and initiator. The activities of the CuI/ligand complexes are highest for tetradentate ligands, lower for tridentate ligands, and lowest for bidentate ligands. Complexes with tripodal and bridged ligands (Me6TREN and bridged cyclam) tend to be more active than those with the corresponding linear ligands.(20)
Addition of the persistent radical, the X-Mtm+1/Ln complex, or its precursors, to the initial reaction medium increases the efficiency of initiation by avoiding the need to form the persistent radical by early stage termination reactions which results in instantaneous polymerization control, higher initiation efficiency and eventually lower cost products.
There are presently several ways to set up the ATRP equilibrium and they will be addressed in greater detail on page 4 of this web site and, where relevant, on various other pages throughout the site.
References
(1) Braunecker, W. A.; Brown, W. C.; Morelli, B. C.; Tang, W.; Poli, R.; Matyjaszewski, K. Macromolecules 2007, 40, 8576-8585.
(2) Wang, J.-S.; Matyjaszewski, K. J. Am. Chem. Soc. 1995, 117, 5614-5615.
(3) Matyjaszewski, K.; Xia, J. Chem. Rev. (Washington, D. C.) 2001, 101, 2921-2990.
(4) Wang, J.-S.; Matyjaszewski, K. Macromolecules 1995, 28, 7901-7910.
(5) Matyjaszewski, K.; Wei, M.; Xia, J.; McDermott, N. E. Macromolecules 1997, 30, 8161-8164.
(6) O'Reilly, R. K.; Gibson, V. C.; White, A. J. P.; Williams, D. J. J. Am. Chem. Soc. 2003, 125, 8450-8451.
(7) O'Reilly, R. K.; Gibson, V. C.; White, A. J. P.; Williams, D. J. Polyhedron 2004, 23, 2921-2928.
(8) Ando, T.; Kamigaito, M.; Sawamoto, M. Macromolecules 2000, 33, 5825-5829.
(9) Uegaki, H.; Kotani, Y.; Kamigaito, M.; Sawamoto, M. Macromolecules 1998, 31, 6756-6761.
(10) Teodorescu, M.; Gaynor, S. G.; Matyjaszewski, K. Macromolecules 2000, 33, 2335-2339.
(11) Gobelt, B.; Matyjaszewski, K. Macromol. Chem. Phys. 2000, 201, 1619-1624.
(12) Kamigaito, M.; Ando, T.; Sawamoto, M. Chem. Rev. (Washington, D. C.) 2001, 101, 3689-3745.
(13) Matyjaszewski, K.; Tsarevsky, N. In PCT Int. Appl.; (Carnegie Mellon University, USA). WO/0228913, 2002; p 64 pp.
(14) Davis, K. A.; Matyjaszewski, K. Journal of Macromolecular Science, Pure and Applied Chemistry 2004, 41, 449-465.
(15) Tang, W.; Matyjaszewski, K. Macromolecules 2007, 40, 1858-1863.
(16) Nicolay, R.; Kwak, Y.; Matyjaszewski, K. Macromolecules 2008, 41, 4585-4596.
(17) Kwak, Y.; Nicolay, R.; Matyjaszewski, K. Macromolecules 2008, 41, 6602-6604.
(18) Huang, C.-F.; Nicolay, R.; Kwak, Y.; Chang, F.-C.; Matyjaszewski, K. Macromolecules 2009, 42, 8198-8210.
(19) Kwak, Y.; Nicolay, R.; Matyjaszewski, K. Macromolecules 2009, 42, 3738-3742.
(20) Tang, W.; Kwak, Y.; Braunecker, W.; Tsarevsky, N. V.; Coote, M. L.; Matyjaszewski, K. J. Am. Chem. Soc. 2008, 130, 10702-10713.
(21) Shen, Y.; Zhu, S. AIChE Journal 2002, 48, 2609-2619.
(22) Jakubowski, W.; Spanswick, J.; Mueller, L.; Matyjaszewski, K. In PCT Int. Appl.; (Carnegie Mellon University, USA). WO 2007075817, 2007; p 65pp.
(23) Braunecker, W. A.; Tsarevsky, N. V.; Gennaro, A.; Matyjaszewski, K. Macromolecules (Washington, DC, United States) 2009, 42, 6348-6360.
(24) Tsarevsky, N. V.; Braunecker, W. A.; Tang, W.; Matyjaszewski, K. ACS Symp. Ser. 2009, 1023, 85-96.
(25) Tang, W.; Nanda, A. K.; Matyjaszewski, K. Macromolecular Chemistry and Physics 2005, 206, 1171-1177.
(26) Tang, W.; Matyjaszewski, K. Macromolecules 2006, 39, 4953-4959.
(27) Matyjaszewski, K.; Wang, J.-S. In PCT Int. Appl.; (Carnegie Mellon University, USA). WO 9630421, 1996; p 129 pp.
(28) Matyjaszewski, K.; Gaynor, S. G.; Coca, S. In PCT Int. Appl.; (Carnegie Mellon University, USA). WO 9840415, 1998; p 230 pp.
(29) Sawamoto, M.; Kamigaito, M. Macromolecular Symposia 2000, 161, 11-18.
(30) Limer, A.; Haddleton, D. M. Macromolecules 2006, 39, 1353-1358.
(31) Haddleton, D. M.; Jarvis, A. P.; Waterson, C.; Bon, S. A. F.; Heming, A. M. ACS Symp. Ser. 2000, 768, 182-196.
(32) Perrier, S.; Haddleton, D. M. Macromolecular Symposia 2002, 182, 261-272.
(33) Lligadas, G.; Ladislaw, J. S.; Guliashvili, T.; Percec, V. Journal of Polymer Science, Part A: Polymer Chemistry 2007, 46, 278-288.
(34) Lligadas, G.; Percec, V. J. Polym. Sci., Part A: Polym. Chem. FIELD Full Journal Title:Journal of Polymer Science, Part A: Polymer Chemistry 2008, 46, 3174-3181.
(35) Bell, C. A.; Whittaker, M. R.; Gahan, L. R.; Monteiro, M. J. J. Polym. Sci., Part A: Polym. Chem. 2007, 46, 146-154.
(36) Matyjaszewski, K.; Tsarevsky, N. V.; Braunecker, W. A.; Dong, H.; Huang, J.; Jakubowski, W.; Kwak, Y.; Nicolay, R.; Tang, W.; Yoon, J. A. Macromolecules 2007, 40, 7795-7806.
(37) Matyjaszewski, K. Macromolecules 1998, 31, 4710-4717.
(38) Matyjaszewski, K.; Gaynor, S. G.; Kulfan, A.; Podwika, M. Macromolecules 1997, 30, 5192-5194.
(39) Tsarevsky, N. V.; Pintauer, T.; Matyjaszewski, K. Macromolecules 2004, 37, 9768-9778.
(40) Matyjaszewski, K.; Tsarevsky, N. In PCT Int. Appl.; (Carnegie Mellon University, USA). WO, 2002; p 64 pp.
(41) Matyjaszewski, K. Macromolecular Symposia 1998, 134, 105-118.
(42) Singleton, D. A.; Nowlan, D. T., III; Jahed, N.; Matyjaszewski, K. Macromolecules 2003, 36, 8609-8616.
(43) Qiu, J.; Matyjaszewski, K.; Thouin, L.; Amatore, C. Macromol. Chem. Phys. 2000, 201, 1625-1631.
(44) Fischer, H. Chemical Reviews 2001, 101, 3581-3610.