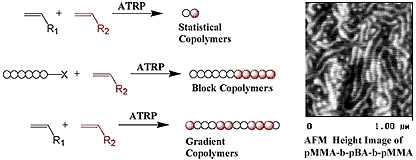
Linear (Co)Polymers
Homopolymers
Linear copolymers
Random or statistical copolymers
Alternating copolymers
Tacticity Control
Homopolymers
There are still some limitations to the range of monomers that can be homopolymerized in an ATRP. The limitation is related to the requirement for repeated reactivation of the dormant species by the transition metal complex. With the current spectrum of catalysts generally there has to be an stabilizing α-substituent adjacent to the transferable atom or group in order that the dormant chain end can be reactivated. The initial range of monomers that could be polymerized by ATRP included styrenes, (meth)acrylates, meth(acrylamides), and acrylonitrile. (2,3) ATRP was then expanded to include several functional monomers including 4-vinyl pyridine,(4) monomers containing an -OH group, such as HEA and HEMA,(5) glycidyl acrylate and precursors of ionic monomers.(6, 7)
Non-polar monomers have also been incorporated into ATRP reactions.(8, 9) See "Starting Points".
The major differences between the polymers prepared by ATRP and prior art polymers prepared by a free radical polymerization are the additional degrees of control over architecture, molecular weight, PDI and telechelic functionality provided by a controlled/"living radical Polymerization (CRP) process.
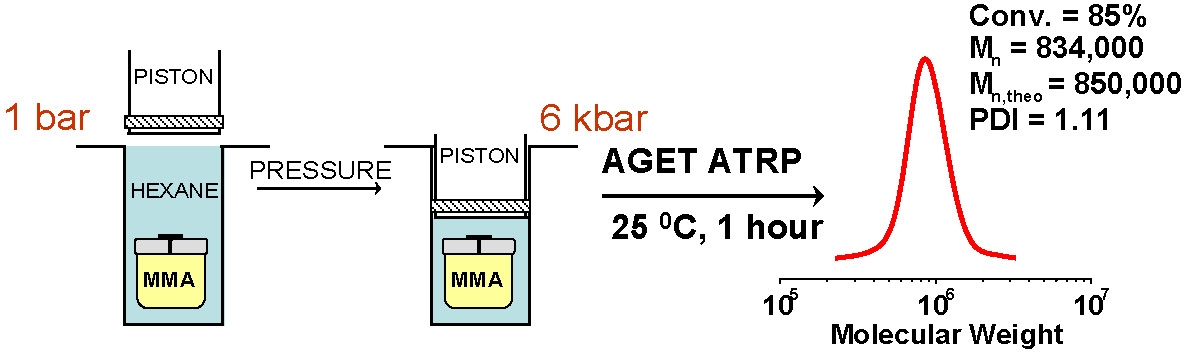
A prior limitation on the maximum molecular weight of the (co)polymers that can be prepared by an ATRP has been surmounted by the development of ARGET ATRP which minimized outer sphere electron transfer (OSET) catalyst based side reactions(10) allowing the production of higher molecular weight polymers, such as polyacrylonitrile, with less apparent tailing.(11) Recent development of ARGET ATRP using a dithiocarbamate initiator was also demonstrated for preparation of ultrahigh molecular weight PMMA.(12)
Another approach to high molecular weight polymers is high pressure. Going to high pressure increases the ratio of kp/kt, therefore propagation is enhanced and termination is repressed and the rate of propagation increased by a few orders of magnitude. For example, at 6 kbar pressure polystyrene with Mn = 1,099,000 g/mol and PDI = 1.24; and methyl methacrylate with Mn = 1,207,000 g/mol and PDI = 1.17 could be synthesized with targeted molecular weight.(13,14)
Linear copolymers
A wide spectrum of copolymers can be prepared via spontaneous or sequential ATRP of two or more monomers with precise control of molar mass, composition and functionality.(15,16) The use of a difunctional initiator allows, for the first time in a radical process, preparation of functional homo-telechelic polymers with almost any desired chain end functionality.(17)
The reactivity ratio of co-monomers in a CRP are very similar to the values found in free radical copolymerization although there are some factors that affect CRP processes with intermittent activation, where differences in repetitive activation or deactivation and suficient time for complete re-equilibration of the system, can result in different rates of consumption of comonomers.(18)
Random or statistical copolymers:
Random copolymers can be prepared by one-pot ATRP of two monomers when there is essentially random incorporation of monomers into the copolymer. This type of copolymer is formed in radical copolymerization when the reactivity ratio of each comonomer is close to one.(13,19) The chemical composition of statistical copolymers prepared by CRP is similar to that formed in a standard free radical copolymerization.(18) Since ARGET ATRP reduces catalyst based side reactions this has also enabled the synthesis of higher molecular weight styrene/acrylonitrile copolymers(20) and improved incorporation of α-olefins into an acrylate copolymerization.(21)
When the reactivity ratios of the comonomers differ and there is preferential incorporation of one monomer in a CRP then spontaneous gradient copolymers are formed in a one pot reaction.
Gradient copolymers:
Gradient copolymers are a special class of copolymer that rose to prominence with the development of ATRP(22-26), since ATRP was the first CRP to allow copolymerization of a range of monomers of differing reactivity.
In contrast to standard radical copolymerization where differences in comonomer reactivity ratios results in variation in instantaneous copolymer composition as the polymerization progresses resulting in the composition of the copolymer formed at the beginning of the copolymerization differing from the composition of copolymers prepared at later stages. In a CRP this variation in the rate of incorporation of the monomers into the copolymer is reflected as a change, or tapering, in composition of the monomer units along the backbone of each copolymer chain.
Gradient copolymers can be prepared by one-pot copolymerization of a selected mole ratio of monomers with sufficiently different reactivity ratios or by controlled feeding of one or more monomers in a manner that forces the composition to be tapered through control over the mole ratio of available monomers, reactivity ratio's still have to be considered.(22-25,27-30) Reports of the preparation of gradient copolymers by iniferter techniques(31) actually led to polymers with broad polydispersity suggesting heterogeneity, since no compositional data was provided.
Gradient copolymers prepared by a CRP differ from the tapered copolymers prepared by living anionic processes(32) which can be described as multi-block copolymers with blocks of different compositions. True gradient copolymers can only be obtained in systems providing fast initiation, uniform chain growth, and easy cross-propagation. Conditions that are difficult to fulfill in ionic systems and earlier radical copolymerization systems.
The shape of the gradient in composition along a polymer backbone can also be changed by use of a difunctional initiator, which leads to a symmetrical "skipping rope" distribution of monomers along the backbone of the copolymer.
Gradient copolymerization can be extended to the formation of graft copolymers with a gradient distribution of grafts, see below.(28)-(33)
Gradient copolymers can also be prepared in biphasic systems by spontaneous copolymerization of appropriate comonomers such as an acrylate and a methacrylate or by controlled addition of one monomer to an active miniemulsion(34) or ab initio ATRP emulsion polymerization where the added comonomer diffuses from the monomer droplets to the active micelle.(35) In addition to cross-propagation kinetics the rate of diffusion of each monomer from the pure comonomer droplet affects the shape of gradient obtained in the final copolymer.Alternating copolymers:
Alternating copolymers may also be prepared by CRP processes. They can be obtained from comonomers that have a spontaneous tendency for alternation: such as copolymerization reactions between a strong electron accepting monomer (maleic anhydride or N-substituted maleimides) and an electron donating monomer (styrene). (8,24,36) Monomers without this inherent tendency towards alternation may also be copolymerized in an alternating fashion by performing CRP in the presence of Lewis acids such as diethyl aluminum chloride or ethyl aluminum sesquichloride. (37-39) Polymerization of complexed monomers is discussed in greater detail in another section of this web page.
REFERENCES
(1) Matyjaszewski, K.; Tsarevsky, N. V. Nature Chemistry 2009, 1, 276-288.
(2) Matyjaszewski, K.; Wang, J.-S. In PCT Int. Appl.; (Carnegie Mellon University, USA). WO 9630421, 1996; p 129 pp.
(3) Matyjaszewski, K.; Jo, S. M.; Paik, H.-j.; Gaynor, S. G. Macromolecules 1997, 30, 6398-6400.
(4) Tsarevsky, N. V.; Braunecker, W. A.; Brooks, S. J.; Matyjaszewski, K. Macromolecules 2006, 39, 6817-6824.
(5) Oh, J. K.; Matyjaszewski, K. Journal of Polymer Science, Part A: Polymer Chemistry 2006, 44, 3787-3796.
(6) Zhang, X.; Matyjaszewski, K. Macromolecules 1999, 32, 1763-1766.
(7) Jakubowski, W.; Matyjaszewski, K. Macromolecular Symposia 2006, 240, 213-223.
(8) Coca, S.; Matyjaszewski, K. Polym. Prepr. 1996, 37, 573-574.
(9) Venkatesh, R.; Klumperman, B. Macromolecules 2004, 37, 1226-1233.
(10) Tsarevsky, N. V.; Braunecker, W. A.; Matyjaszewski, K. Journal of Organometallic Chemistry 2007, 692, 3212-3222.
(11) Dong, H.; Tang, W.; Matyjaszewski, K. Macromolecules 2007, 40, 2974-2977.
(12) Kwak, Y.; Matyjaszewski, K. Macromolecules 2008, 41, 6627-6635.
(13) Arita, T.; Kayama, Y.; Ohno, K.; Tsujii, Y.; Fukuda, T. Polymer FIELD Full Journal Title:Polymer 2008, 49, 2426-2429.
(14) Kwiatkowski, P.; Jurczak, J.; Pietrasik, J.; Jakubowski, W.; Mueller, L.; Matyjaszewski, K. Macromolecules 2008, 41, 1067-1069.
(15) Gaynor, S. G.; Matyjaszewski, K. ACS Symp. Ser. 1998, 685, 396.
(16) Qin, S.; Saget, J.; Pyun, J.; Jia, S.; Kowalewski, T.; Matyjaszewski, K. Macromolecules 2003, 36, 8969-8977.
(17) Shipp, D. A.; Wang, J.-L.; Matyjaszewski, K. Macromolecules 1998, 31, 8005-8008.
(18) Matyjaszewski, K. Macromolecules 2002, 35, 6773-6781.
(19) Davis, K. A.; Matyjaszewski, K. Advances in Polymer Science 2002, 159, 2-166.
(20) Pietrasik, J.; Dong, H.; Matyjaszewski, K. Macromolecules 2006, 39, 6384-6390.
(21) Tanaka, K.; Matyjaszewski, K. Macromolecules 2007, 40, 5255-5260.
(22) Greszta, D.; Matyjaszewski, K. Polymer Preprints 1996, 37, 569-570.
(23) Matyjaszewski, K.; Ziegler, M. J.; Arehart, S. V.; Greszta, D.; Pakula, T. J. Phys. Org. Chem. 2000, 13, 775-786.
(24) Matyjaszewski, K.; Coca, S.; Gaynor, S. G.; Greszta, D.; Patten, T. E.; Wang, J.-s.; Xia, J. In PCT Int. Appl.; (Carnegie Mellon University, USA). WO199718247, 1997; p 182 pp.
(25) Matyjaszewski, K.; Coca, S.; Gaynor, S. G.; Greszta, D.; Patten, T. E.; Wang, J.-S.; Xia, J. In USPTO; Carnegie Mellon University: US6887962, 2005.
(26) Greszta, D., 1997; p 276 pp.
(27) Matyjaszewski, K.; Coca, S.; Gaynor, S. G.; Greszta, D.; Patten, T. E.; Wang, J.-S.; Xia, J. In USPTO; CMU: US5807937, 1998.
(28) Lee, H.-i.; Matyjaszewski, K.; Yu, S.; Sheiko, S. S. Macromolecules 2005, 38, 8264-8271.
(29) Farcet, C.; Charleux, B.; Pirri, R. Macromolecular Symposia 2002, 182, 249-260.
(30) Boerner, H. G.; Duran, D.; Matyjaszewski, K.; da Silva, M.; Sheiko, S. S. Macromolecules 2002, 35, 3387-3394.
(31) Kuchanov, S. I.; Olenin, A. V. Vysokomolekulyarnye Soedineniya, Seriya B: Kratkie Soobshcheniya 1991, 33, 563-564.
(32) Hashimoto, T.; Tsukahara, Y.; Tachi, K.; Kawai, H. Macromolecules 1983, 16, 648-657.
(33) Lord, S. J.; Sheiko, S. S.; LaRue, I.; Lee, H.-I.; Matyjaszewski, K. Macromolecules 2004, 37, 4235-4240.
(34) Min, K. E.; Li, M.; Matyjaszewski, K. Journal of Polymer Science, Part A: Polymer Chemistry 2005, 43, 3616-3622.
(35) Min, K.; Oh, J. K.; Matyjaszewski, K. Journal of Polymer Science, Part A: Polymer Chemistry 2007, 45, 1413-1423.
(36) Davies, M. C.; Dawkins, J. V.; Hourston, D. J. Polymer 2005, 46, 1739-1753.
(37) Kirci, B.; Lutz, J.-F.; Matyjaszewski, K. Macromolecules 2002, 35, 2448-2451.
(38) Lutz, J.-F.; Kirci, B.; Matyjaszewski, K. Macromolecules 2003, 36, 3136-3145.
(39) Denizli, B. K.; Lutz, J.-F.; Okrasa, L.; Pakula, T.; Guner, A.; Matyjaszewski, K. Journal of Polymer Science, Part A: Polymer Chemistry 2005, 43, 3440-3446.
Tacticity control:
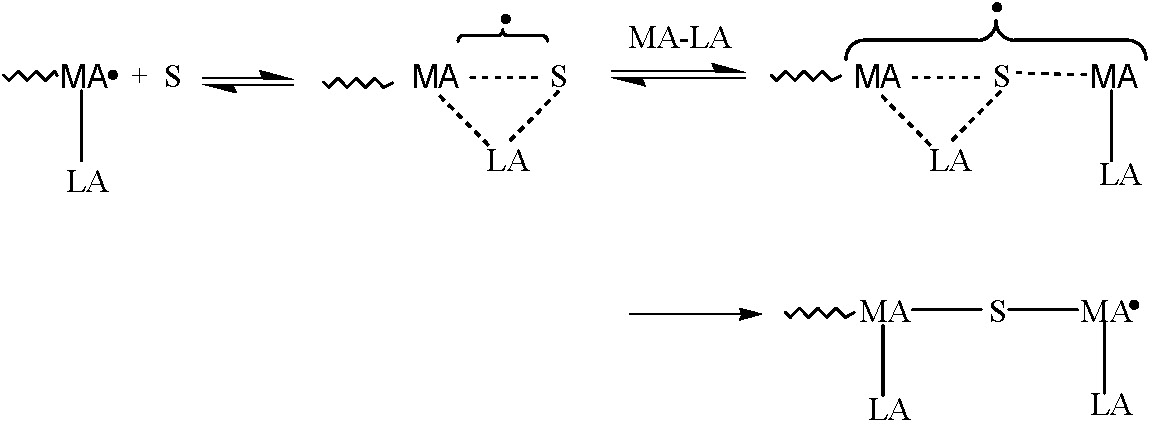
Until recently CRP has been less successful at attaining control over chain microstructure in terms of sequence distribution and tacticity since, due to the radical nature of the propagation step, the chemoselectivity (reactivity ratios), regioselectivity (proportions of head to head units) and stereoselectivity (tacticity) of polymers formed in a CRP are similar to those in conventional radical polymerization. Recently the controlled alternating copolymerization of a donor monomer (styrene) and an acceptor monomer (alkyl methacrylate or alkyl acrylate) complexed with a Lewis acid was examined using several CRP processes, including ATRP, NMP, iodide degenerative transfer polymerization (IDTP), and RAFT.
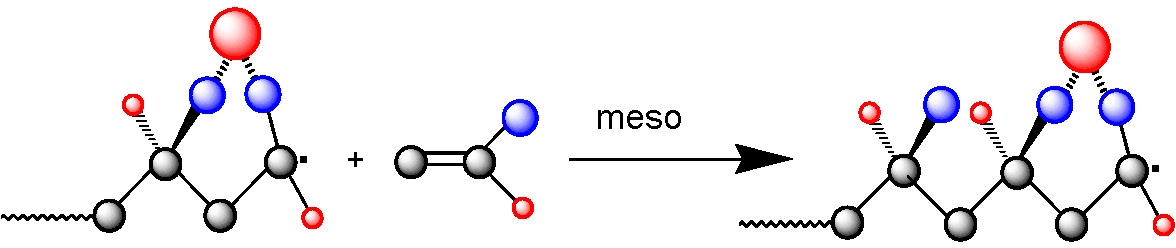
Complex formation increases the electron-accepting ability of the acceptor monomer, which strongly enhances the cross-propagation rate constants and thereby increases the tendency for alternating copolymerization. RAFT polymerization was found to be the most versatile system.(1) The combination of RAFT and Lewis acids complexation techniques allowed synthesis of well-defined poly(styrene-alt-methyl methacrylate) copolymers with controlled molecular weight (up to Mn = 70,000 g mol-1), low polydispersities (Mw/Mn < 1.3), and controlled comonomer sequences (approx.90% of alternating triads).
These results were obtained in the presence of diethylaluminum chloride and ethylaluminum sesquichloride. Moreover, the alternating copolymers obtained in this reaction retain chain end functionality and were used as macroinitiators for the synthesis of well-defined diblock copolymers poly(methyl methacrylate-alt-styrene)-b-polystyrene.(2)
Generally, free radical addition reactions or homolytic chain propagation reactions are not stereoselective. However Okamoto determined that addition of Lewis acids can provide stereocontrol in the radical polymerization of acrylic monomers.(3,4) As noted above, conditions for CRP in the presence of Lewis acids had been developed. Therefore, all three major controlled/living radical polymerization processes, ATRP, RAFT and NMP, were investigated for the polymerization of N,N-dimethylacrylamide in the presence of Lewis acids known to enhance isotacticity, such as yttrium trifluoromethanesulfonate (Y(OTf)3) and ytterbium trifluoromethanesulfonate (Yb(OTf) 3).(5)
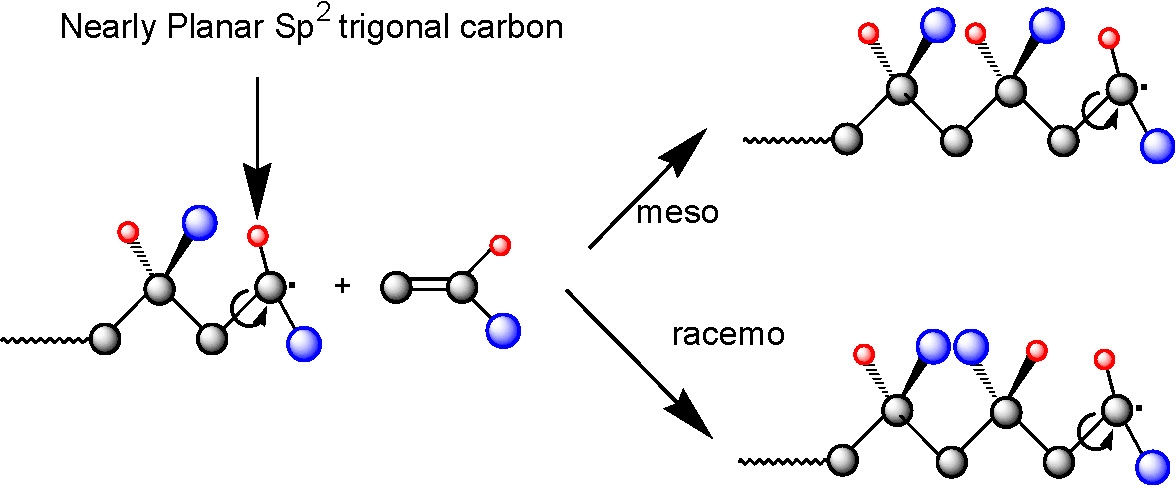
Poly(N,N-dimethylacrylamide) with controlled MW, low polydispersity, and a high proportion of meso dyads (approx.85%) was prepared using ATRP (methyl 2-chloropropionate/CuCl/Me6TREN) and RAFT (with cumyl dithiobenzoate transfer agent) in the presence of Y(OTf)3. The combination of NMP (using N-tert-butyl-1-diethylphosphono-2,2-dimethylpropyl nitroxide, SG1) and a Lewis acid complexation technique led to less precise control over chain architecture and microstructure (approximately 65% meso dyads), than RAFT/Y(OTf)3 or ATRP/Y(OTf)3. The latter two systems were used for the first one-pot synthesis of stereoblock copolymers by radical polymerization. Well-defined stereoblock copolymers, atactic-b-isotactic poly(N,N-dimethylacrylamides), were obtained by adding Y(OTf)3 to either an ongoing RAFT or ATRP polymerization, started in the absence of the Lewis acid.(6)
RAFT polymerization of MMA in the presence of of cyanoisopropyl Me trithiocarbonate [CH3SC(:S)SC(CH3)2CN] and scandium triflate Sc(OTf)3 provided simultaneous well controlled RAFT polymerization and Lewis acid-controlled tacticity.(7)
This was later extended to iron catalyzed polymerization of acrylamides in the presence of Lewis acids then to the use of triple hydrogen bonding for stereospecific polymerization of a DAD monomer which provided simultaneous control of tacticity and molecular weight.(8)
Indeed in a recent review article Kamigaito (9) provided a summary of the requirements for stereocontrol in a CRP. The stereo-regulation should rely on an added Lewis acid, a polar solvent, or a multiple hydrogen-bonding additive, which can interact with the polar groups around the growing polymer terminal units and/or the monomer to induce the stereospecific chain growth, while the living or controlled chain growth is governed by a metal catalyst or a reversible chain transfer agent. The stereospecific living radical polymerization is therefore achievable by the judicious combination of the stereocontrol additive or solvent and the living radical polymerization catalyst or mediator. This methodology is relatively easily applied to the precise synthesis of stereoblock or stereogradient polymers, in which the stereochemistry of the growing chain is either abruptly or gradually changed, respectively.
REFERENCES
(1) Kirci, B.; Lutz, J.-F.; Matyjaszewski, K. Macromolecules 2002, 35, 2448-2451.
(2) Lutz, J.-F.; Kirci, B.; Matyjaszewski, K. Macromolecules 2003, 36, 3136-3145.
(3) Okamoto, Y.; Habaue, S.; Isobe, Y.; Nakano, T. Macrom. Symp. 2002, 183, 83-88.
(4) Sugiyama, Y.; Satoh, K.; Kamigaito, M.; Okamoto, Y. Journal of Polymer Science, Part A: Polymer Chemistry 2006, 44, 2086-2098.
(5) Lutz, J.-F.; Neugebauer, D.; Matyjaszewski, K. Journal of the American Chemical Society 2003, 125, 6986-6993.
(6) Lutz, J.-F.; Jakubowski, W.; Matyjaszewski, K. Macromolecular Rapid Communications 2004, 25, 486-492.
(7) Chong, Y. K.; Moad, G.; Rizzardo, E.; Skidmore, M. A.; Thang, S. H. Macromolecules (Washington, DC, United States) 2007, 40, 9262-9271.
(8) Wan, D.; Satoh, K.; Kamigaito, M. Macromolecules 2006, 39, 6882-6886.
(9) Kamigaito, M.; Satoh, K. Macromolecules 2008, 41, 269-276.