Graft Copolymers
Grafting through
Grafting from
Grafting to
Graft copolymers belong to the general class of segmented copolymers and generally consist of a linear backbone of one composition and randomly distributed branches of a different composition. They have been prepared for many decades and have been used as impact resistant materials, thermoplastic elastomers, compatibilizers or emulsifiers for the preparation of stable blends or alloys. The number of potential applications has now expanded with the development of CRP.
Well-defined graft copolymers are most frequently prepared by either a) a "grafting through" or b) a "grafting from" controlled polymerization process. However the development of "click" chemistry(1) has led to a third approach, c) based on site specific "grafting to" chemistry.
a) "Grafting through"
The "grafting through" method (or macromonomer method) is one of the simplest ways to synthesize graft copolymers with well defined side chains.
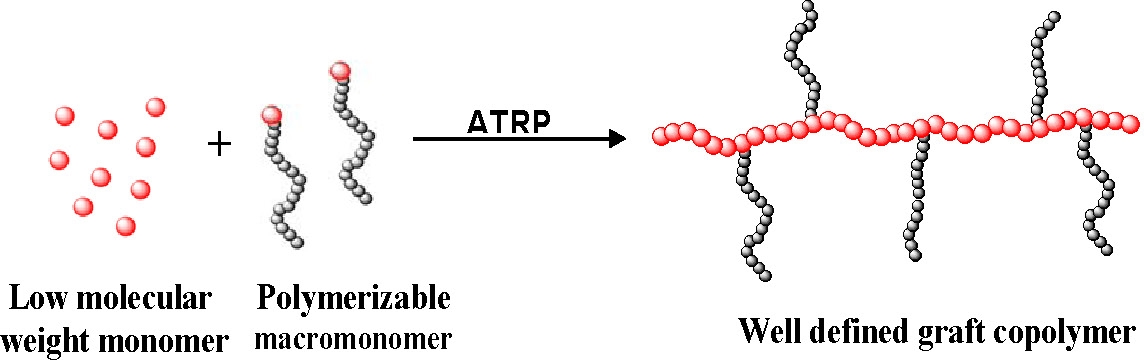
Typically a low molecular weight monomer is radically copolymerized with a (meth)acrylate functionalized macromonomer. This method permits incorporation of macromonomers that have prepared by other controlled polymerization processes into a backbone prepared by a CRP. Macromonomers such as polyethylene,(2,3) poly(ethylene oxide),(4) polysiloxanes,(5) poly(lactic acid),(6) polycaprolactone(7) have been incorporated into a polystyrene or poly(meth)acrylate backbone.
Moreover, it is possible to design well-defined graft copolymers by combining the CRP "grafting through" macromonomers procedure where the macromonomers had been prepared by any controlled polymerization process.(8) This combination of controlled polymerization processes allows control of polydispersity, functionality, copolymer composition, backbone length, branch length and branch spacing by consideration of mole-ratio of the MM in the feed and reactivity ratio of both the monomer and macromonomer. Branches can be distributed homogeneously or heterogeneously based on the reactivity ratio of the terminal functional group on the macromonomer and the low molecular weight monomer; and, as shown in the properties section, this has a significant effect on the physical properties of the materials.(6,7)
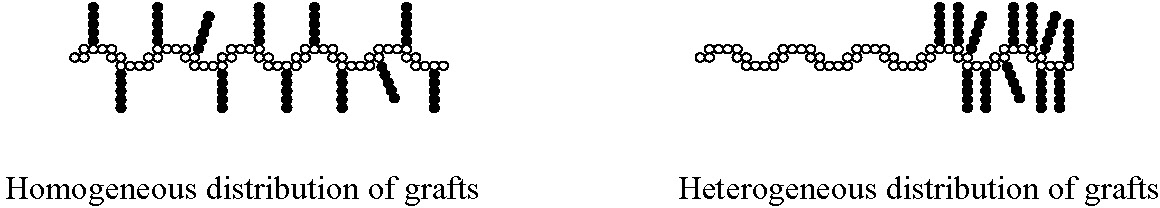
A series of segmented poly(alkyl methacrylate)-g-poly(D-lactide)/poly(dimethylsiloxane) terpolymers, with different topologies were prepared by employing a combination of the "grafting through" technique and CRP.(9) Two synthetic pathways were used. The first was a single-step approach in which a methacrylate monomer (methyl methacrylate or butyl methacrylate) was copolymerized with a mixture of a PLA macromonomer and a PDMS macromonomer. The second strategy was a two-step approach in which a graft copolymer containing one macromonomer was chain-extended by a copolymerization of the second macromonomer and the low-molecular weight methacrylate monomer. The molecular structure of the terpolymers was investigated by 2D GPC which indicated that well-defined terpolymers with controlled branch distribution were prepared via both pathways. The topologies of the graft terpolymers prepared by different combinations of the two step approach are displayed in the following schematic diagrams.
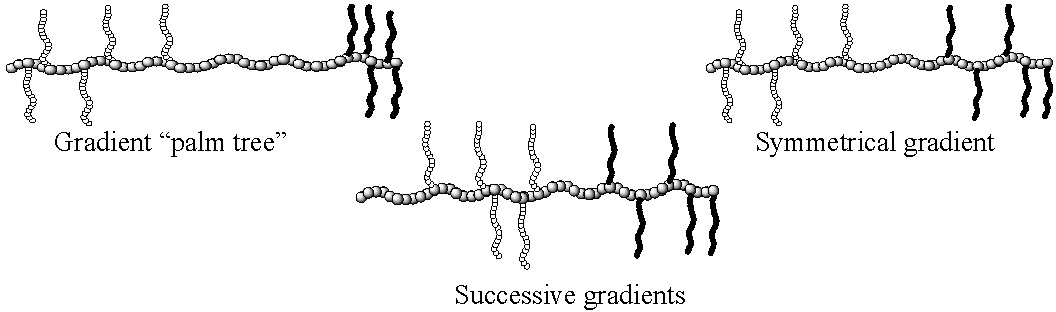
While properties have yet to be fully explored, it is likely that phase separation is modified by polymer topology and hence the properties of the material will differ even though compositions were held constant.
A spontaneous gradient graft copolymer was prepared by grafting through two different PEO macromonomers.(10) Selection of a PEO methacrylate with a methyl end-group (PEOMeMA, DP of the PEO = 23) and a PEO acrylate end-capped by a phenyl ring (PEOPhA, DP of the PEO = 4) for the copolymerization led to a spontaneous gradient of PEO grafts along the copolymer backbone. A gradient copolymer was formed because of the significant difference in the reactivity of the two PEO macromonomers. The resulting copolymer has a high fraction of PEOMeMA grafts at one end of the polymer chain, gradually changing through hetero-sequences to predominately PEOPhA at the other chain end. An increase in the initial feed ratio of PEO acrylate reduced the rate of change in the shape of the gradient.
b) "Grafting from"
The primary requirement for a successful "grafting from" reaction is a preformed macromolecule with distributed initiating functionality.
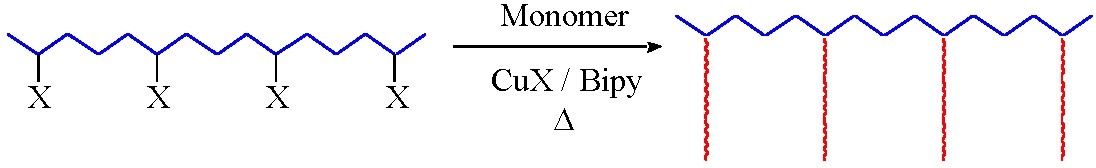
Grafting from reactions have been conducted from polyethylene,(11-13) polyvinylchloride,(14,15) and polyisobutylene.(16,17) The only requirement for a multifunctional ATRP grafting from macroinitiator is distributed radically transferable atoms along the polymer backbone. The initiating sites can be incorporated by copolymerization,(11,14) be an inherent part of the first polymer,(15) or incorporated in a post-polymerization reaction.(16) Indeed a pre-functionalized isobutylene rubber is commercially available from Exxon, (EXXPRO 3035) and has been employed in a grafting from ATRP of MMA forming a spectrum of materials ranging from elastomers to impact resistant poly(MMA).(16)
Densely grafted copolymers are also prepared by a "grafting from" polymerization and are discussed in the section on bottle-brush macromolecules and on the "nanocomposites" page.
c) "Grafting to"
Grafting to has become a more efficient method for the preparation of graft copolymers with the rise of various "click" chemistries. This approach has been used for the preparation of well defined star molecules,(18) loosely grafted copolymers(19) and as noted in the following section densely grafted structures.(20, 21)
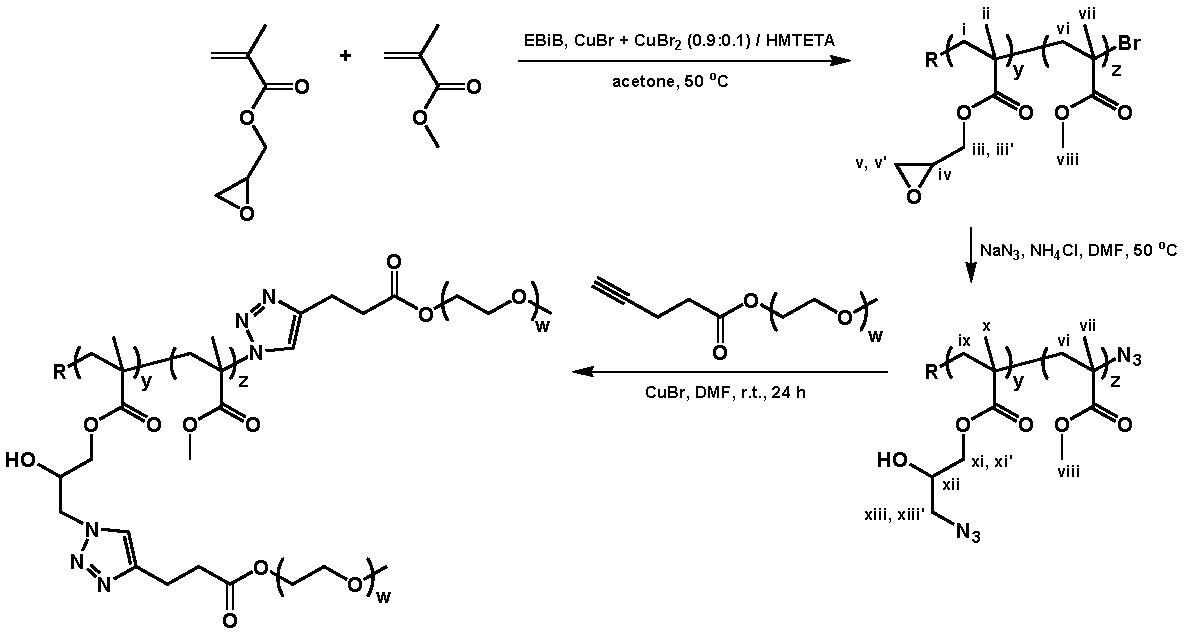
A well-defined copolymers of glycidyl methacrylate (<40 mol %) and Me methacrylate prepared by ATRP. The glycidyl butyrate monomer unit that had been incorporated into the copolymer was efficiently opened with sodium azide in the presence of ammonium chloride in DMF at 50 0C to prepare a copolymer suitable for "grafting to" using click linking chemistry. This click-type reaction led to the formation of a copolymer with distributed units of the corresponding 1-hydroxy-2-azido functional group in high yields. These azide-cont aining copolymers were further functionalized in a second click reaction conducted at room temp. CuBr/N,N,N',N'',N''-pentamethyldiethylenetriamine-catalyzed 1,3-dipolar cyclo-addition of poly(ethylene oxide) methyl ether pentynoate yielded loosely grafted polymeric brushes with hydrophilic PEO side chains.
Another "high yield" chemical reaction, familiar to all interested in CRP, was exploited in a simple one-pot synthesis of heterograft copolymers via "grafting onto" by atom transfer nitroxide radical coupling chemistry.(22) The main chain was prepared by anionic ring-opening copolymerization of ethylene oxide (EO) and 4-glycidyloxy-2,2,6,6-tetramethylpiperidine-1-oxyl (GTEMPO) then polystyrene and poly(tert-Bu acrylate) with bromine end (PS-Br, PtBA-Br) were prepared by ATRP. When the three polymers were mixed in the presence of CuBr/N,N,N',N'',N''-pentamethyldiethylenetriamine (PMDETA) at 90 0C, the formed secondary carbon radicals at the PS and PtBA chain ends were quickly trapped by nitroxide radicals on poly(GTEMPO-co-EO). It was found that the density of GTEMPO groups on the main chain of the poly(GTEMPO-co-EO), the MW of PS/PtBA side chains, and the structure of macroradicals significantly affected the efficiency of the grafting to reaction.
Heterograft copolymers poly(4-glycidyloxy-2,2,6,6-tetramethylpiperidine-1-oxyl-co- ethylene oxide)-graft-polystyrene and poly(tert-Bu acrylate) (poly(GTEMPO-co-EO)-g-PS/PtBA) were synthesized in one-pot by atom transfer nitroxide radical coupling (ATNRC) reaction via "graft onto."
REFERENCES
(1) Kolb, H. C.; Finn, M. G.; Sharpless, K. B. Angewandte Chemie, International Edition 2001, 40, 2004-2021.
(2) Hong, S. C.; Jia, S.; Teodorescu, M.; Kowalewski, T.; Matyjaszewski, K.; Gottfried, A. C.; Brookhart, M. Journal of Polymer Science, Part A: Polymer Chemistry 2002, 40, 2736-2749.
(3) Kaneyoshi, H.; Inoue, Y.; Matyjaszewski, K. Macromolecules 2005, 38, 5425-5435.
(4) Neugebauer, D.; Zhang, Y.; Pakula, T.; Sheiko, S. S.; Matyjaszewski, K. Macromolecules 2003, 36, 6746-6755.
(5) Shinoda, H.; Matyjaszewski, K.; Okrasa, L.; Mierzwa, M.; Pakula, T. Macromolecules 2003, 36, 4772-4778.
(6) Shinoda, H.; Matyjaszewski, K. Macromolecules 2001, 34, 6243-6248.
(7) Hawker, C. J.; Mecerreyes, D.; Elce, E.; Dao, J.; Hedrick, J. L.; Barakat, I.; Dubois, P.; Jerome, R.; Volksen, i. Macromol. Chem. Phys. 1997, 198, 155-166.
(8) Matyjaszewski, K.; Beers, K. L.; Kern, A.; Gaynor, S. G. J. Polym. Sci., Part A: Polym. Chem. 1998, 36, 823-830.
(9) Lutz, J.-F.; Jahed, N.; Matyjaszewski, K. Journal of Polymer Science, Part A: Polymer Chemistry 2004, 42, 1939-1952.
(10) Neugebauer, D.; Zhang, Y.; Pakula, T. Journal of Polymer Science, Part A: Polymer Chemistry 2006, 44, 1347-1356.
(11) Inoue, Y.; Matsugi, T.; Kashiwa, N.; Matyjaszewski, K. Macromolecules 2004, 37, 3651-3658.
(12) Okrasa, L.; Pakula, T.; Inoue, Y.; Matyjaszewski, K. Colloid and Polymer Science 2004, 282, 844-853.
(13) Kaneyoshi, H.; Inoue, Y.; Matyjaszewski, K. PMSE Preprints 2004, 91, 41-42.
(14) Paik, H. J.; Gaynor, S. G.; Matyjaszewski, K. Macromol. Rapid Commun. 1998, 19, 47-52.
(15) Percec, V.; Asgarzadeh, F. J. Polym. Sci., Part A: Polym. Chem. 2001, 39, 1120-1135.
(16) Hong, S. C.; Pakula, T.; Matyjaszewski, K. Macromolecular Chemistry and Physics 2001, 202, 3392-3402.
(17) Matyjaszewski, K.; Gaynor, S. G.; Coca, S. In PCT Int. Appl.; (Carnegie Mellon University, USA). WO 9840415, 1998; p 230 pp.
(18) Gao, H.; Min, K.; Matyjaszewski, K. Macromolecular Chemistry and Physics 2007, 208, 1370-1378.
(19) Tsarevsky, N. V.; Bencherif, S. A.; Matyjaszewski, K. Macromolecules 2007, 40, 4439-4445.
(20) Sumerlin, B. S.; Tsarevsky, N. V.; Louche, G.; Lee, R. Y.; Matyjaszewski, K. Macromolecules 2005, 38, 7540-7545.
(21) Gao, H.; Matyjaszewski, K. Journal of the American Chemical Society 2007, 129, 6633-6639.
(22) Fu, Q.; Liu, C.; Lin, W.; Huang, J. J. Polym. Sci., Part A: Polym. Chem. 2008, 46, 6770-6779.