Post-polymerization modification of monomer units
Advantages:
-
Allows incorporatation of functionality that is incompatible with the polymerization process
-
Allows characterization of the initial copolymer prior to further functionalization
-
Facilitates "grafting to" or "grafting from" reactions
Post-polymerization modification of incorporated monomer units focus on two types of reaction.
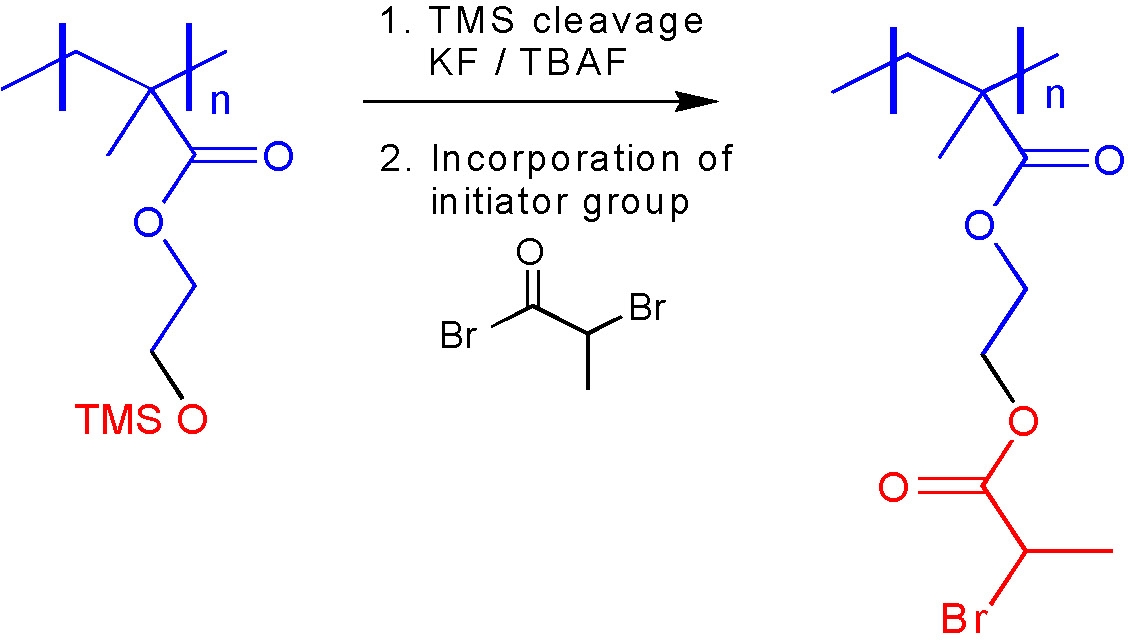
One is the removal of protecting groups where monomers with the desired functionality are incompatible with one or more components of the selected polymerization process. The functional monomers are polymerized with a protected functional group that is deprotected to provide the desired functionality after the reaction is complete. Examples include acidic monomers such as (meth)acrylic acid, isomeric vinylbenzoic acids, or unsaturated sulfonic or phosphonic acids that protonate and therefore "destroy" the transition metal complexes with N-based ligands typically used as ATRP catalysts. Although some moderately successful attempts have been made to polymerize MAA by ATRP, in general, protected acids are the preferred approach. Examples of protective groups include t-Bu(1,2) for acrylic acid functionality or the TMS group frequently used as a protective group for the precursor of brush macromolecules.(3)
The other approach is to copolymerize monomers with one functional group then convert that functional group into the desired functional group after the first polymerization is complete.
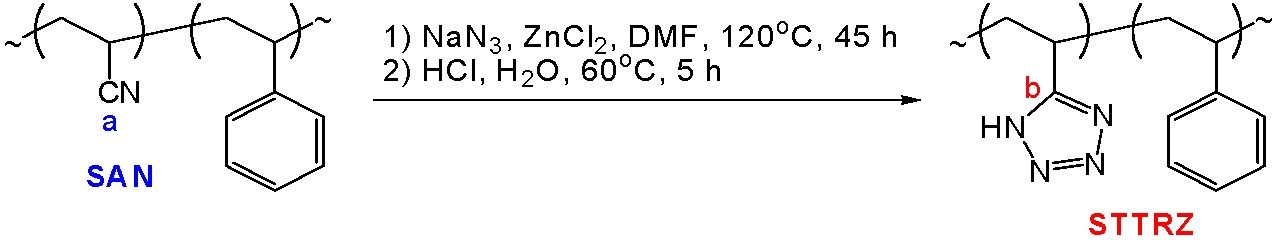
One example of a post-polymerization reaction to form distributed acid functionality is formation of copolymers with distributed tetrazole units. Tetrazoles are both acidic, resembling carboxylic acids, and coordinating compounds and hence the direct ATRP of vinyltetrazoles has not been reported. The preparation of well-defined tetrazole-containing polymers can be accomplished using ATRP to (co)polymerize AN followed by a "click" chemistry(4,5) chemical modification of the produced polyAN to yield the desired materials.(6)
A number of high yield "click" chemistry functionalization of monomer units in a copolymer backbone have been conducted as shown below.(4,5,7-9)
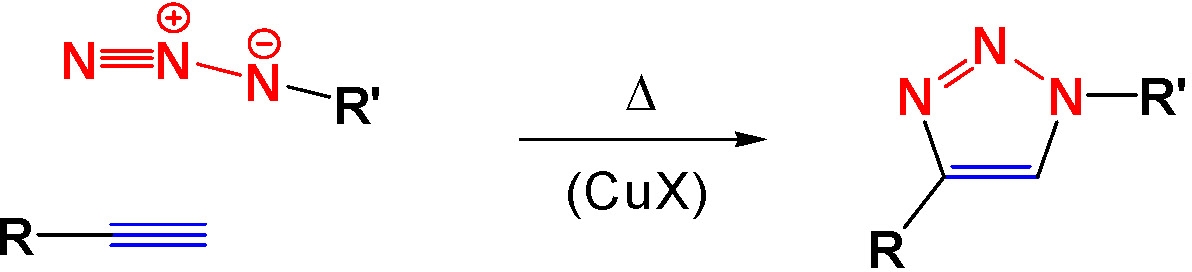
In another example a novel monomer, 3-azidopropyl methacrylate (AzPMA), was polymerized via ATRP with good control of the polymer molecular weight distribution and retention of chain functionality.(8) Poly(3-azidopropyl methacrylate) was coupled with propargyl alcohol, propargyl triphenylphosphonium bromide, propargyl 2-bromoisobutyrate, and 4-pentynoic acid via a highly efficient "click" reactions in the presence of a CuI catalyst. The azido-functionalized polymer demonstrated enhanced reactivity during "click" reactions compared to small molecules with comparable structures. The ability of the coupling reactions to be conducted at room temperature without significant excess of reagents makes this an attractive alternative approach for the preparation of (co)polymers with high degrees of functionalization.(8)
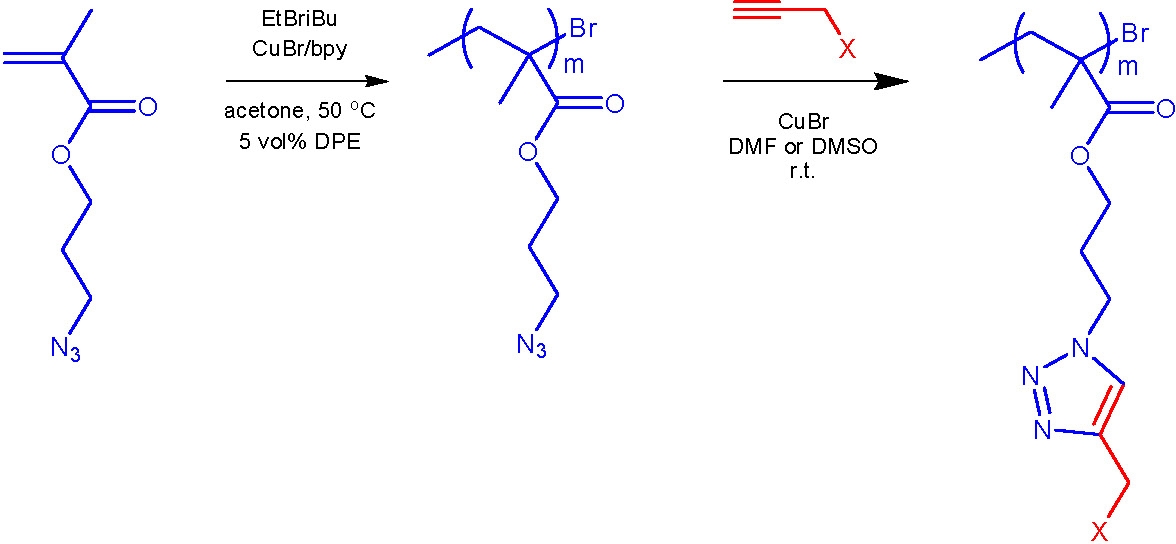
To avoid synthesis of the heat- and shock-sensitive azide-containing monomer, azido groups were introduced in polymers containing epoxide groups using a click-type ring-opening reaction. The azide-containing polymers thus prepared were further modified using alkyne-azide-type click reactions.(10)
As mentioned above, HEA and HEMA(3,11) are easy to (co)polymerize by ATRP, and the polymers containing hydroxyl-groups that can be used in esterification reactions with various functionalized carboxylic acids to attach a different functional groups to the polymer backbone. This approach is frequently used to prepare multifunctional macroinitiators for the preparation of bottle-brush copolymers.
However, esterifications are not always efficient, especially when a polymeric substrate is used. Alternatively poly(glycidyl acrylate) copolymers can be prepared by ATRP to serve as a precursor of functional polymers, since the pendant glycidyl group can react with nucleophiles and thereby serve as a precursor of functional polymers.(12)
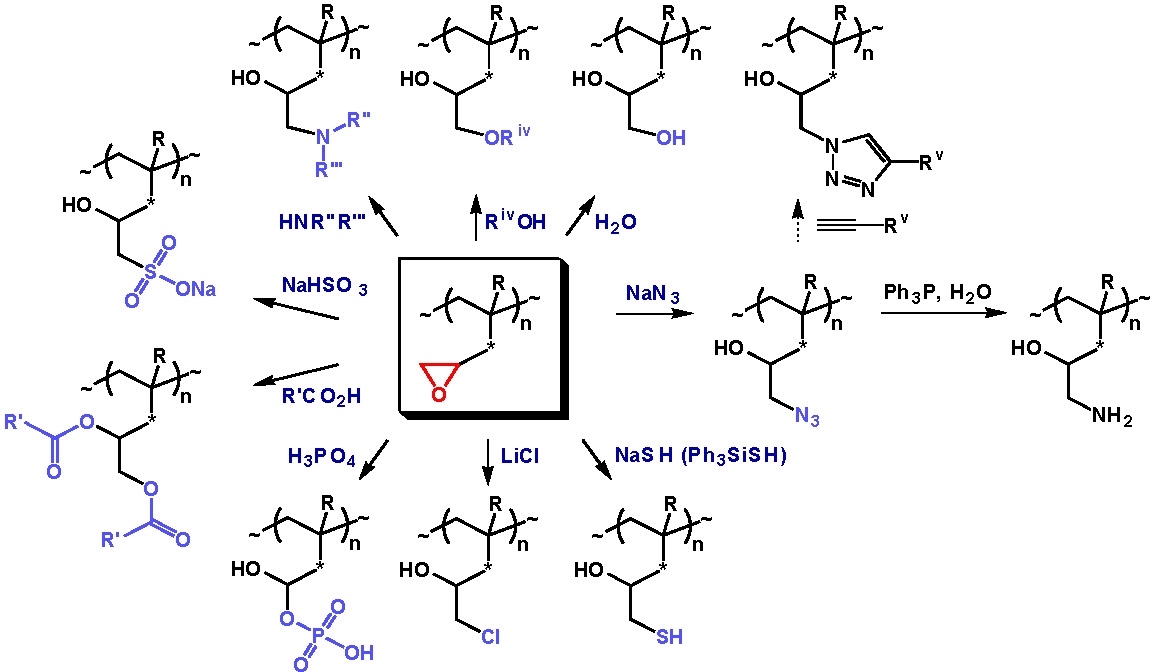
REFERENCES
(1) Davis, K. A.; Matyjaszewski, K. Macromolecules 2000, 33, 4039-4047.
(2) Davis, K. A.; Charleux, B.; Matyjaszewski, K. J. Polym. Sci., Part A: Polym. Chem. 2000, 38, 2274-2283.
(3) Beers, K. L.; Boo, S.; Gaynor, S. G.; Matyjaszewski, K. Macromolecules 1999, 32, 5772-5776.
(4) Huisgen, R. 1,3 [One,Three]-Dipolar Cycloaddit. Chem. 1984, 1, 1-176.
(5) Kolb, H. C.; Finn, M. G.; Sharpless, K. B. Angewandte Chemie, International Edition 2001, 40, 2004-2021.
(6) Tsarevsky, N. V.; Bernaerts, K. V.; Dufour, B.; Du Prez, F. E.; Matyjaszewski, K. Macromolecules 2004, 37, 9308-9313.
(7) Wu, P.; Feldman, A. K.; Nugent, A. K.; Hawker, C. J.; Scheel, A.; Voit, B.; Pyun, J.; Frechet, J. M. J.; Sharpless, K. B.; Fokin, V. V. Angewandte Chemie, International Edition 2004, 43, 3928-3932.
(8) Sumerlin, B. S.; Tsarevsky, N. V.; Louche, G.; Lee, R. Y.; Matyjaszewski, K. Macromolecules 2005, 38, 7540-7545.
(9) Golas, P. L.; Tsarevsky, N. V.; Matyjaszewski, K. Macromol. Rapid Commun. 2008, 29, 1167-1171.
(10) Tsarevsky, N. V.; Bencherif, S. A.; Matyjaszewski, K. Macromolecules 2007, 40, 4439-4445.
(11) Coca, S.; Jasieczek, C. B.; Beers, K. L.; Matyjaszewski, K. J. Polym. Sci., Part A: Polym. Chem. 1998, 36, 1417-1424.
(12) Tsarevsky, N. V.; et al.; NSTI Nanotech, Nanotechnology Conference and Trade Show, Technical Proceedings, Boston, MA, United States, June 1-5, 2008, 2008, 2, 665-668.