Normal ATRP
The procedure for "normal" initiation of an ATRP reaction employs an initiator molecule, either a small molecule, macromolecule, or a functionalized surface, with one or more transferable atoms or groups, most often a (pseudo)halide that undergoes a one electron redox reaction with a transition metal catalyst that is added to the reaction medium in a lower oxidation state forming the reactive species and the catalyst complex in the higher oxidation state capable of deactivating the propagating radical.(1-7)
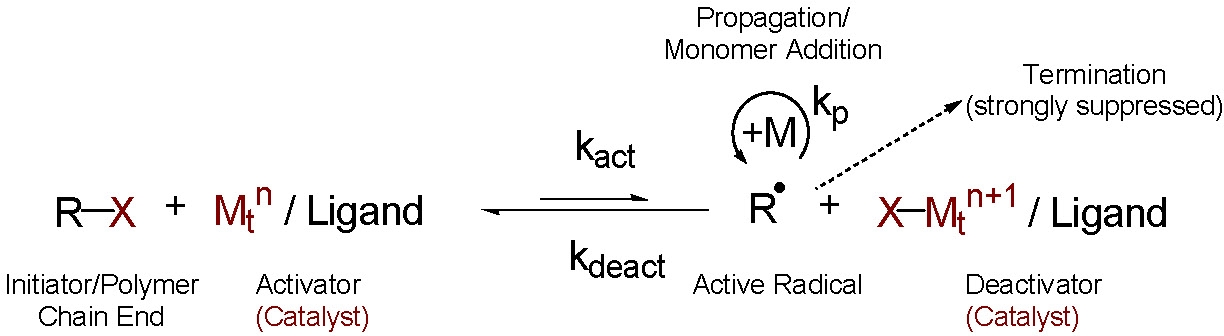
In all of the published literature on ATRP this R-X molecule has been called the initiator, even though in contrast to standard free radical polymerization initiators, this molecule is an inherently thermally stable entity and is incorporated in its entirety into the final polymer. Therefore the complete "initiator" molecule is an integral part of the final material. The added initiator, R-X, can be a mono functional initiator or a multifunctional initiator; i.e. it can possess more than one functional group capable of providing a site for chain growth. The added initiator can also be used to introduce additional functionality into the α-chain end of a linear copolymer or within the "core" of a multi-armed star or composite material. It can be a macroinitiator (a polymer containing one or more initiator site(s)) or the initiator(s) can be attached to any type of surface, either a particle, a flat surface, a fiber, or a porous material.
The transition metal catalyst (Mtn/L), where Mtn is the transition metal in the lower oxidation state n complexed with appropriate ligand(s) L, reacts reversibly with the added initiator molecule to transfer the radically transferable atom or group from the initiator to generate an oxidized transition metal halide complex (X-Mtn+1/L) and a radical (R·). This radical propagates, adding monomer (M), and is rapidly deactivated by reaction with the oxidized transition metal halide complex to reform the lower oxidation state transition metal catalyst and an oligomeric X-terminated chain (R-P1+-X).
This sequence can repeat itself, until the desired level of consumption of the monomer is reached, resulting in the synthesis of polymers with predetermined molecular weights (DPn = D[M]/[RX]0) and low polydispersities (Mw/Mn < 1.5).
The primary advantage of a "normal ATRP initiation" procedure is that it provides great freedom for the choice of both initiator and catalyst complex. The most active catalyst can be selected (provided all reagents are pure and oxygen free) resulting in lower levels of transition metal and other impurities in the final product. However care has to be taken selecting the appropriate initiator, catalyst and reaction conditions since activation rate constants kact for a variety of initiators can vary by a factor of one million.(7,8)
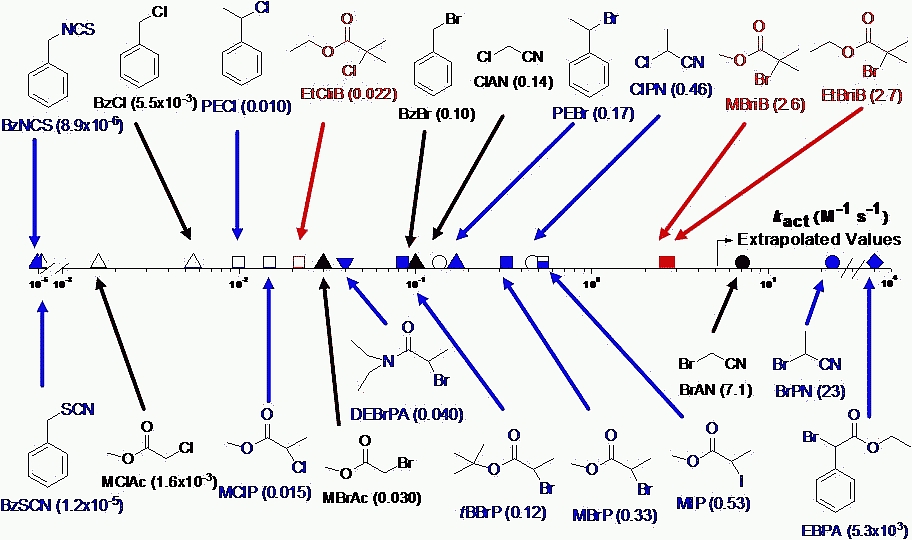
Plot taken from Wei Tang's presentation at the CRP Consortium Meeting October 2006
In small scale reactions care should be taken to reduce the amount of dissolved oxygen in the system, since oxygen will oxidize the catalyst complex forming the higher oxidation state transition metal complex, the redox conjugate, which is the equivalent of the persistent radical in an ATRP, and reduce the rate of reaction.(9,10)
Note however that the addition of a low level of the redox conjugate to the initial reaction medium does provide complete control from the start of the polymerization and increases the amount of added initiator incorporated into the final product, by reducing the fraction of termination reactions between low molecular weight species normally required to form the persistent radical.(6) A study on the kinetics of normal ATRP with addition of 20% CuII versus CuI showed that the ATRP equilibrium was reached almost immediately and the molecular weight of the polymers were closer to theoretical values and polymers with lower dispersity were obtained.(8) This is important in industrial scale processes where the cost of a functional initiator may be a major concern.
The selection of the appropriate radically transferable atom or group on the initiator and transition metal complex is important to provide control over the efficiency of the initiation reaction.(11,12)
REFERENCES
(1) Wang, J.-S.; Matyjaszewski, K. J. Am. Chem. Soc. 1995, 117, 5614-5615.
(2) Kato, M.; Kamigaito, M.; Sawamoto, M.; Higashimura, T. Macromolecules 1995, 28, 1721-1723.
(3) Percec, V.; Barboiu, B. Macromolecules 1995, 28, 7970-7972.
(4) Granel, C.; Dubois, P.; Jerome, R.; Teyssie, P. Macromolecules 1996, 29, 8576-8582.
(5) Matyjaszewski, K.; Wang, J.-S. In PCT Int. Appl.; (Carnegie Mellon University, USA). WO 9630421, 1996; p 129 pp.
(6) Matyjaszewski, K.; Coca, S.; Gaynor, S. G.; Greszta, D.; Patten, T. E.; Wang, J.-s.; Xia, J. In PCT Int. Appl.; (Carnegie Mellon University, USA). WO199718247, 1997; p 182 pp.
(7) Tang, W.; Matyjaszewski, K. Macromolecules 2007, 40, 1858-1863.
(8) Tang, W.; Matyjaszewski, K. Macromol. Theory Simul. 2008, 17, 359-375.
(9) Patten, T. E.; Matyjaszewski, K. Adv. Mater. 1998, 10, 901-915.
(10) Fischer, H. J. Polym. Sci., Part A: Polym. Chem. 1999, 37, 1885-1901.
(11) Gillies, M. B.; Matyjaszewski, K.; Norrby, P.-O.; Pintauer, T.; Poli, R.; Richard, P. Macromolecules 2003, 36, 8551-8559.
(12) Matyjaszewski, K.; Qin, S.; Boyce, J. R.; Shirvanyants, D.; Sheiko, S. S. Macromolecules 2003, 36, 1843-1849.