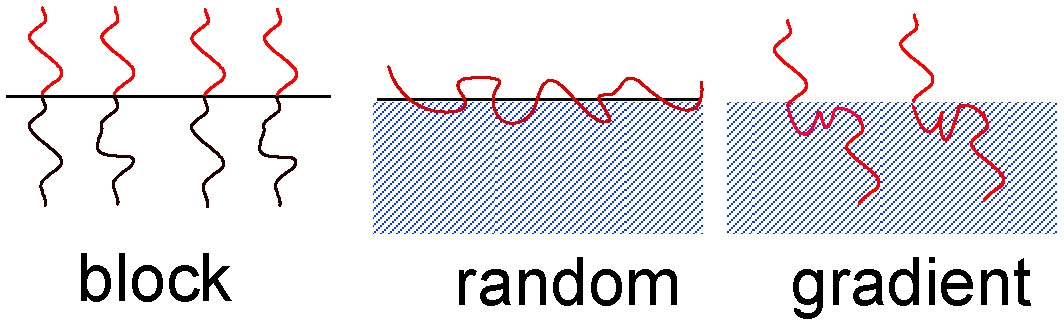
Linear copolymers
Alternating and Random
Gradient Copolymers
Gradient Block Copolymers
Linear Copolymers
Another objective of the Matyjaszewski group is to interact with other research groups to develop an understanding of the properties of materials prepared for the first time by CRP. However, since the properties of the materials prepared by CRP can be "adjusted" by many subtle and presently unexplored ways, we use the word "evaluation" in the title of this section to indicate that the study is by no means complete. We hope the following brief overview might give you some ideas on how to construct materials with the spectrum of properties required by your target application.
Many of the following slides were provided by Professor Tadeusz Pakula from the Max-Planck-Institute for Polymer Research, Mainz, Germany who had collaborated with the group for over a decade. The slides are taken from a presentation made at the March 2005 CRP Consortium Meeting. The slides have been selected to show how the bulk properties of a material depend on the structure, size scales and heterogeneity of the material, and the dynamics, or time scales, of the test, and how the response rates of the polymer to stress ultimately depend on molecular structure. They illustrate how different molecular elements on a macroscopic continuum are affected by the physics of processing and how they interact with the chemical parameters controlled by synthesis. They are presented here in order to provide a foundation from which to understand or learn from many of the figures presented later on this page, that are discussed in greater detail in papers co-authored by Professor Pakula and Professor Matyjaszewski. The first slide illustrates how the macroscopic continuum of phase separation in a polymer can be influenced at different levels by physical or chemical manipulation of the material.
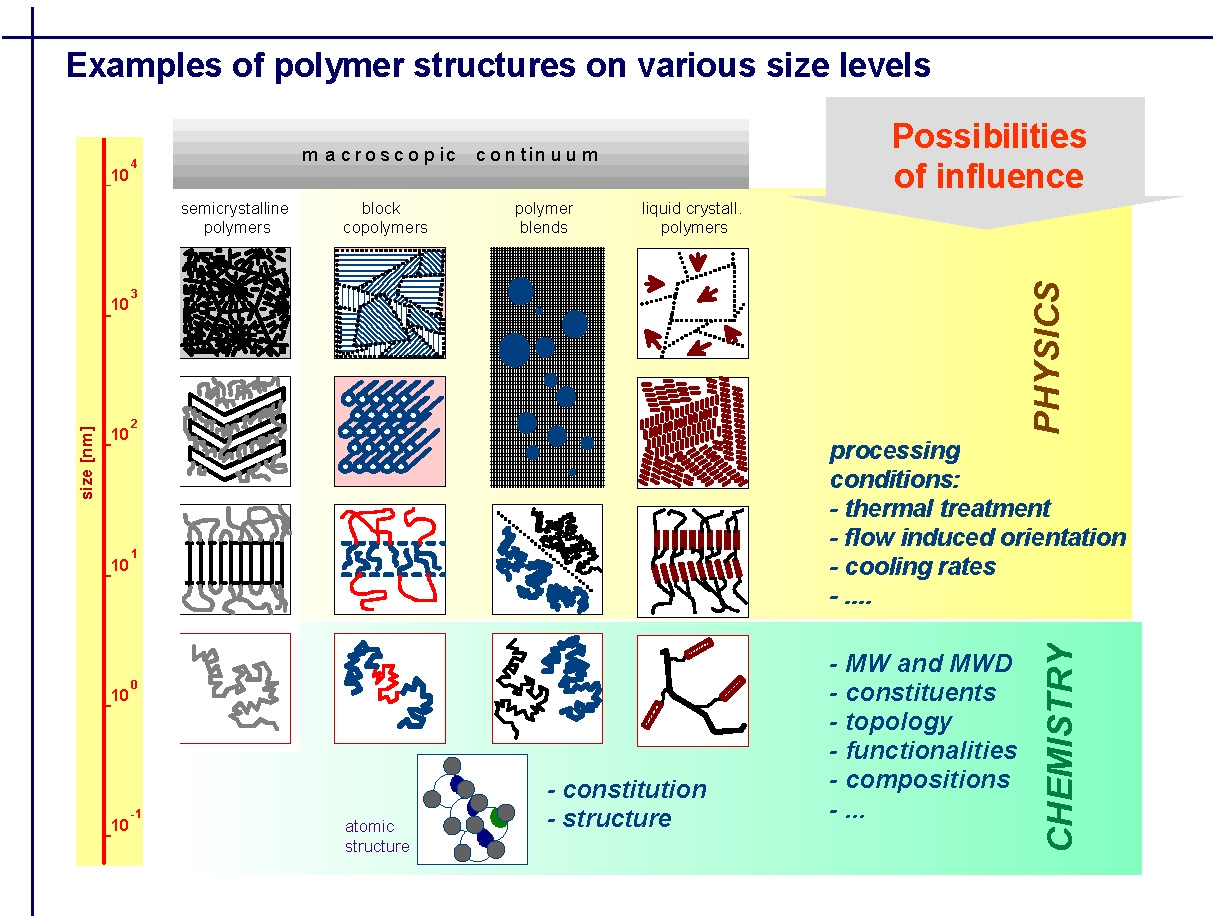
The next slide illustrates how the physical properties of a material are dependent on the composition or morphological elements of the material and how the scale of the dynamics of molecular motion change with temperature.

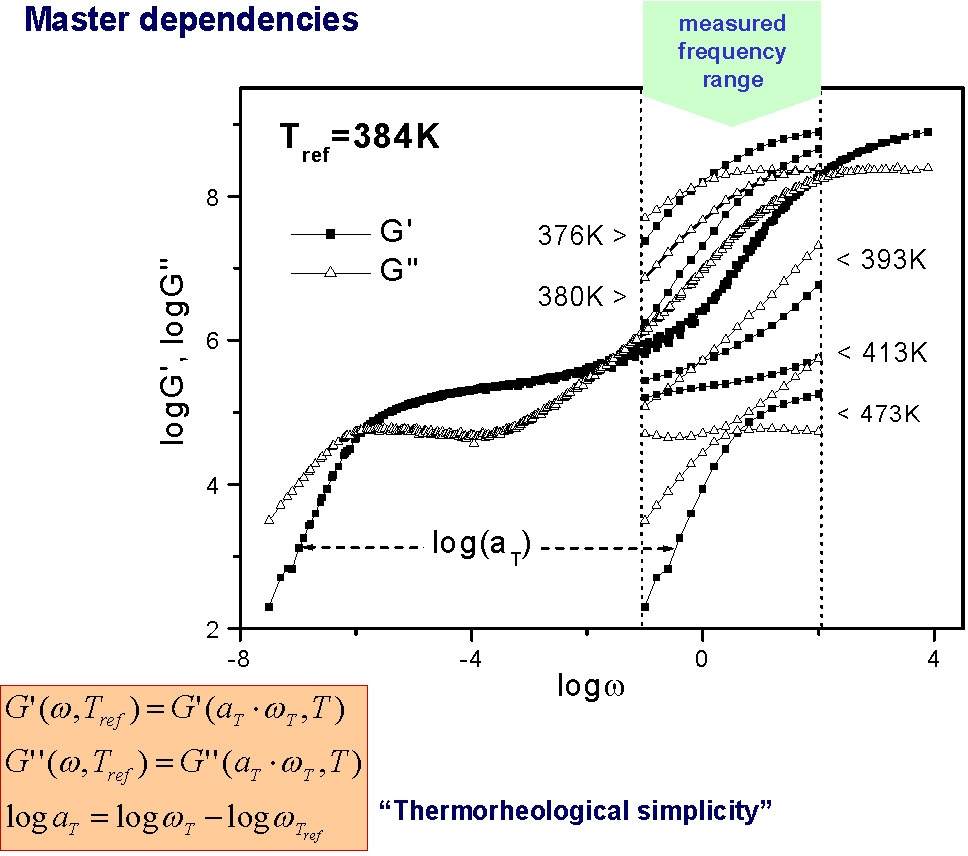
In many of the following figures, changes in polymer dynamics over several orders of magnitude have been measured using mechanical spectroscopy. The final spectrum is constructed from mechanical spectroscopy measured over a fairly narrow frequency range at different temperatures. When the curves are fitted together they provide an illustration of mechanical response over a wide range of physical environments.
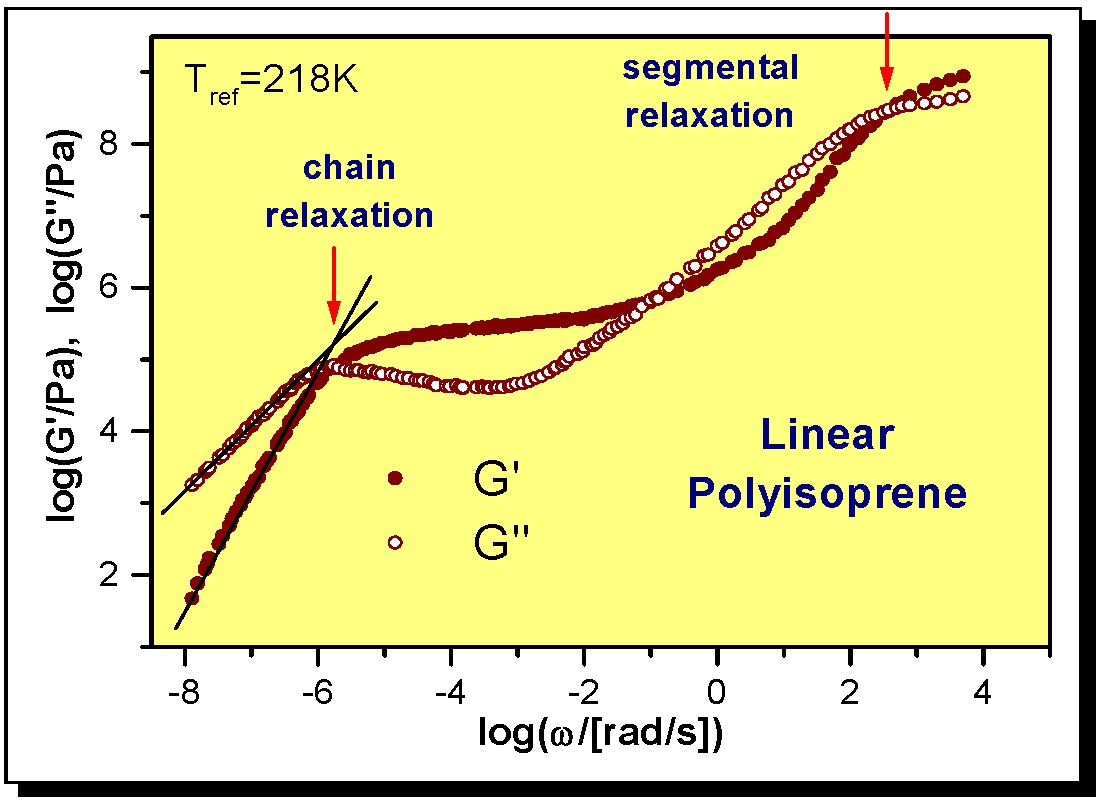
The following figure shows the spectrum of a linear polyisoprene.
The figure clearly shows the transition from the glassy state through segmental relaxation, transitioning into a rubbery state, seen as a plateau, prior to chain relaxation and polymer flow.
Professor Pakula has done a great deal of work on modeling such systems, particularly Monte Carlo and Molecular Dynamics during his interactions with a spectrum of synthetic chemists around the world, but we will focus solely on the results obtained from materials uniquely prepared by CRP at CMU
There are a multiplicity of materials that could be discussed in each of the following topical sub-headings but we will focus on a very few examples that were selected in the hope they show how materials prepared by CRP differ from materials prepared using earlier polymerization processes.
Linear copolymers
Alternating and random copolymers:
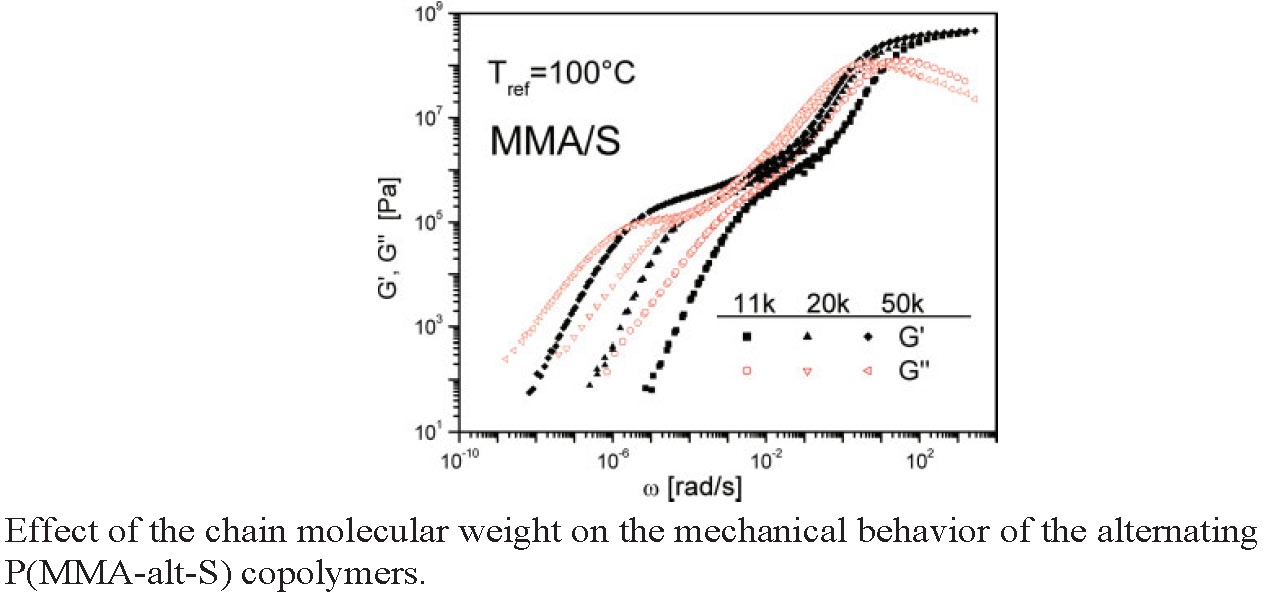
The physical properties of two well-defined alternating copolymers, poly(Me methacrylate-alt-styrene) and poly(Bu methacrylate-alt-styrene), prepared by reversible addition-fragmentation chain transfer polymerization in the presence of Lewis acids, were investigated with differential scanning calorimetry, wide-angle X-ray scattering, and dynamic mechanical measurements. The properties were compared the corresponding homopolymers and with those of random copolymers of the same overall composition. Wide-angle X-ray scattering data showed that the alternating copolymers possessed a more regular comonomer sequence than the random copolymers. The thermo-mechanical properties of alternating copolymers and random copolymers were quite similar and, as expected, typical for amorphous polymers but in one of the cases studied, the butyl methacrylate-alt-styrene copolymer, the glass transition temperature for alternating copolymer was markedly higher than the random copolymer; 41.7 Vs. 29.2 °C for the alternating and random copolymers respectively.(1)
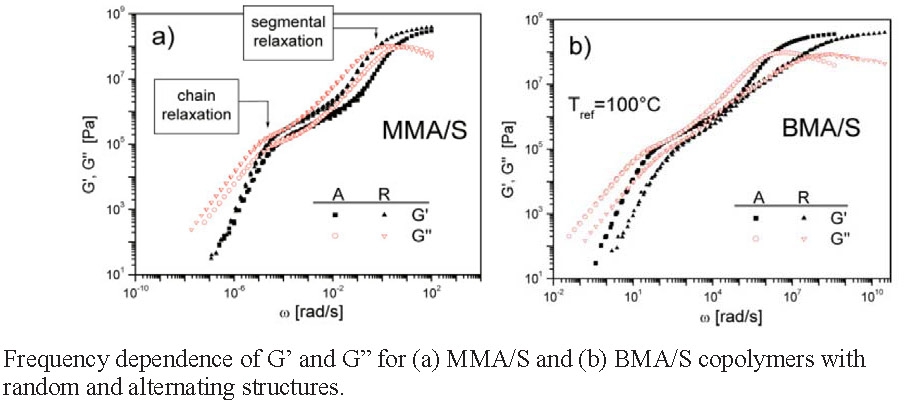
Small changes in the molecular weight result in changes in the segmental relaxation, meaning that chain relaxation rate is strongly dependent on the molecular weight, and an increase in the molecular weight results in an extension of the range of the rubbery behavior.
Gradient Copolymers:
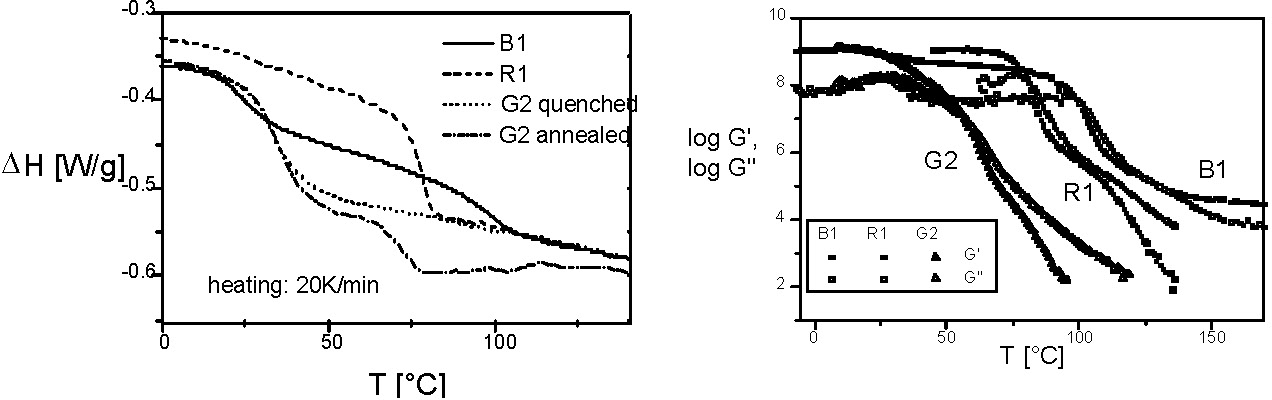
Gradient copolymers could not be prepared until a robust Controlled Radical Copolymerization (CRP) was developed.(2) Until recently the(3) cross-propagation kinetics in "living" ionic processes did not allow synthesis of such materials. The initial paper on properties of gradient copolymers(4) provides a comparison between the properties of a styrene-methyl acrylate gradient copolymer (G2), a random copolymer (R1) and a block (B1) copolymer, each with the same overall composition. The following figures show the DSC thermograms and the temperature dependencies of the storage (G') and loss moduli (G'') for the three materials. The extraordinary broad temperature range for the segmental relaxation in the gradient copolymer is due to a wide spectrum of compositions within the chain.
It is possible to manipulate the phase transitions in a gradient copolymer by controlling the molecular weight, composition and shape of the gradient along the polymer backbone. Potential applications for gradient copolymers include reinforcing agents, blend compatibilizers, where properties are influenced by the higher Critical Micelle Concentrations (CMC) of gradient copolymers compared to block copolymers,(5-7) or, because their behavior at an interface can be controlled, as impact modifiers and sound or vibration dampeners. In addition, because of the ability to prepare materials with a broad and selectively adjustable range of Tg's, gradient copolymers will find application as pressure sensitive adhesives, and wetting or leveling additives for coatingsor inks.(8,9)
In a paper discussing the "Cooperative Motion Algorithm"(10) Professor Pakula indicated that smoothing of the chain-end distributions are most efficient for gradient copolymers. A later joint paper demonstrated that the volume fraction of a gradient copolymer that resides within the interphase of a phase separating copolymer can be modified by changing the gradient along the polymer backbone resulting in a very broad segmental relaxation. Consequentially the composition changes continuously across the interphasial boundary and the volume fraction of the material within the boundary layer can be controlled thereby modifying the bulk properties of the material.(11)
Gradient block copolymers:
As a transition to the next section, focusing on block copolymers, the following figures are taken from reference 4 and show how the properties of an ABA block copolymer can be changed by incorporation of a low concentration of a second monomer into the A blocks. The polymers are GM13, a clean ABA block copolymer (MMA131-BA507-MM A131), and GM14, a copolymer with a fraction of the B-monomer in the A-blocks (MMA115/BA15)-BA527-(MMA115/BA15). The presence of a gradient of composition along the backbone of the A-blocks leads to a block copolymer with reduced ultimate stress but much higher ultimate elongation than the comparable block copolymer with pure MMA A-blocks.
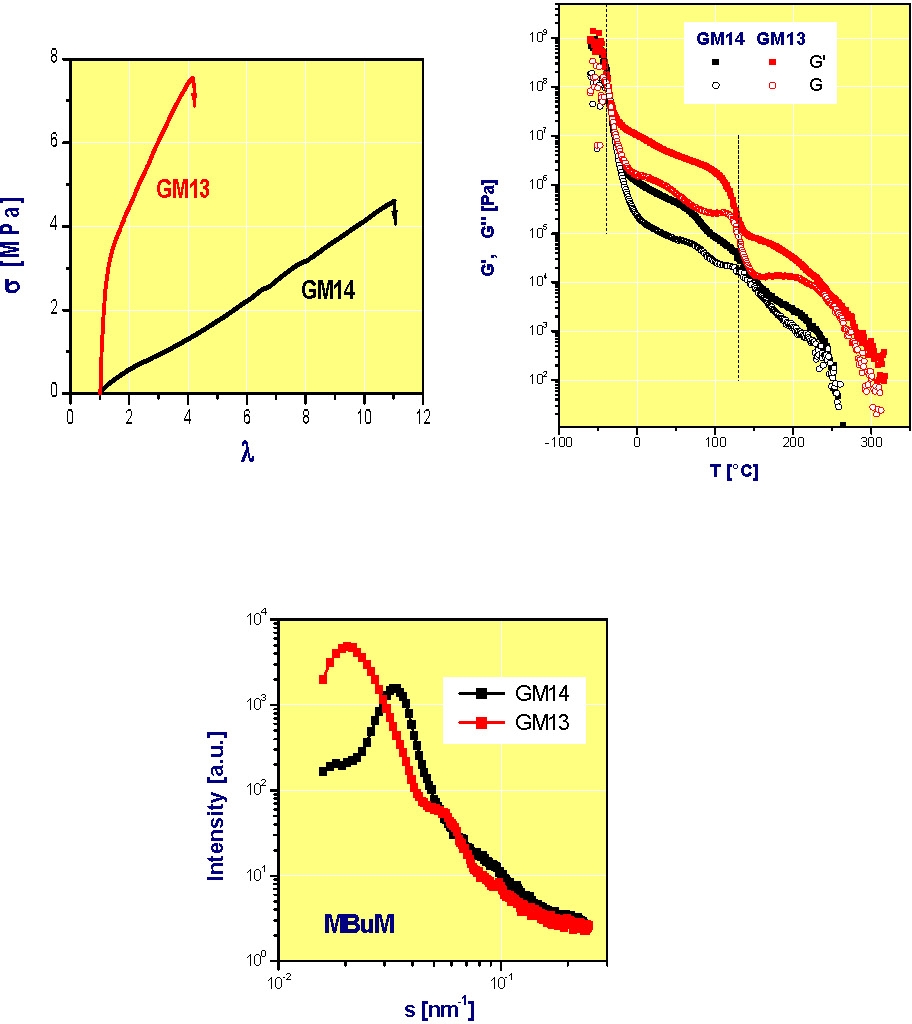
The reason for this behavior is that a higher mole fraction of the A block copolymer segments reside in the interphasial layer resulting in smaller domains of the "A" blocks for G14 than G13 even though both block copolymers have the same number of MMA units in each A-block, compared to the pure block copolymer. Differences in solubility behavior of block vs. gradient block have also been reported.(12)
REFERENCES
(1) Denizli, B. K.; Lutz, J.-F.; Okrasa, L.; Pakula, T.; Guner, A.; Matyjaszewski, K. Journal of Polymer Science, Part A: Polymer Chemistry 2005, 43, 3440-3446.
(2) Greszta, D., 1997; p 276 pp.
(3) Park, J.-S.; Kataoka, K. Macromolecules 2007, 40, 3599-3609.
(4) Matyjaszewski, K.; Ziegler, M. J.; Arehart, S. V.; Greszta, D.; Pakula, T. J. Phys. Org. Chem. 2000, 13, 775-786.
(5) Wang, T.; Liu, F.; Luo, N.; Ying, S.; Liu, Q. Hecheng Xiangjiao Gongye 2001, 24, 328-330.
(6) Kim, J.; Zhou, H.; Nguyen, S. T.; Torkelson, J. M. Polymer 2006, 47, 5799-5809.
(7) Wong, C. L. H.; Kim, J.; Roth, C. B.; Torkelson, J. M. Macromolecules 2007, 40, 5631-5633.
(8) Goebelt, B. FATIPEC Congress 2004, 27th, 303-312.
(9) Kim, J.; Mok, M. M.; Sandoval, R. W.; Woo, D. J.; Torkelson, J. M. Macromolecules 2006, 39, 6152-6160.
(10) Pakula, T. Macromolecular Theory and Simulations 1996, 5, 987-1006.
(11) Lutz, J.-F.; Pakula, T.; Matyjaszewski, K. ACS Symposium Series 2003, 854, 268-282.
(12) Vidts, K. R. M.; Dervaux, B.; Du Prez, F. E. Polymer 2006, 47, 6028-6037.