Molecular Assembly of Block Copolymers
Controlling morphology by PDI:
Multisegmented blocks:
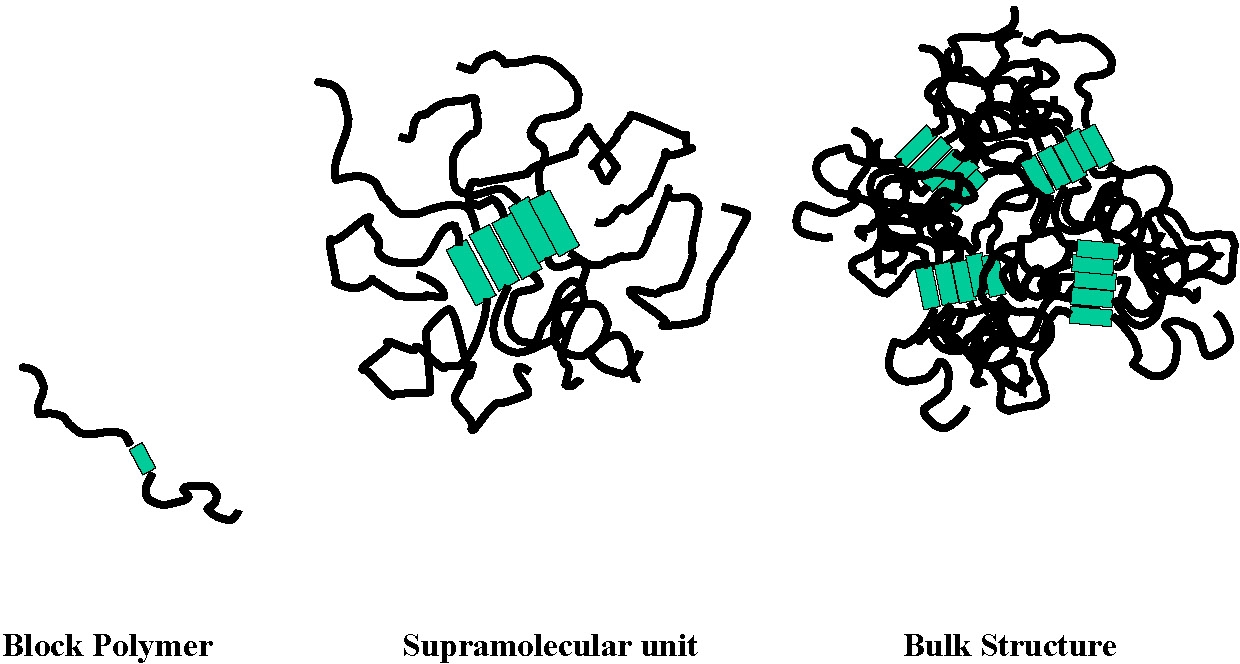
A multiplicity of novel block copolymers can be synthesized as a result of the development of CRP. Amongst them are a number of novel materials prepared by dual polymerization mechanisms such as an homotelechelic macroinitiator prepared by a polycondensation process followed by ATRP.(1) Rather unexpectedly the bulk morphology of an ABA tri-block copolymer with a polysulfone B block and two butylacrylate A-blocks led to the development unique properties. The simple schematic of a block copolymer, and even its phase separation into two separate phases of known dimensions, disguises the repercussions of the formation of a supramolecular structural unit that impacts the bulk structure, and hence the physical properties of the material.(2)
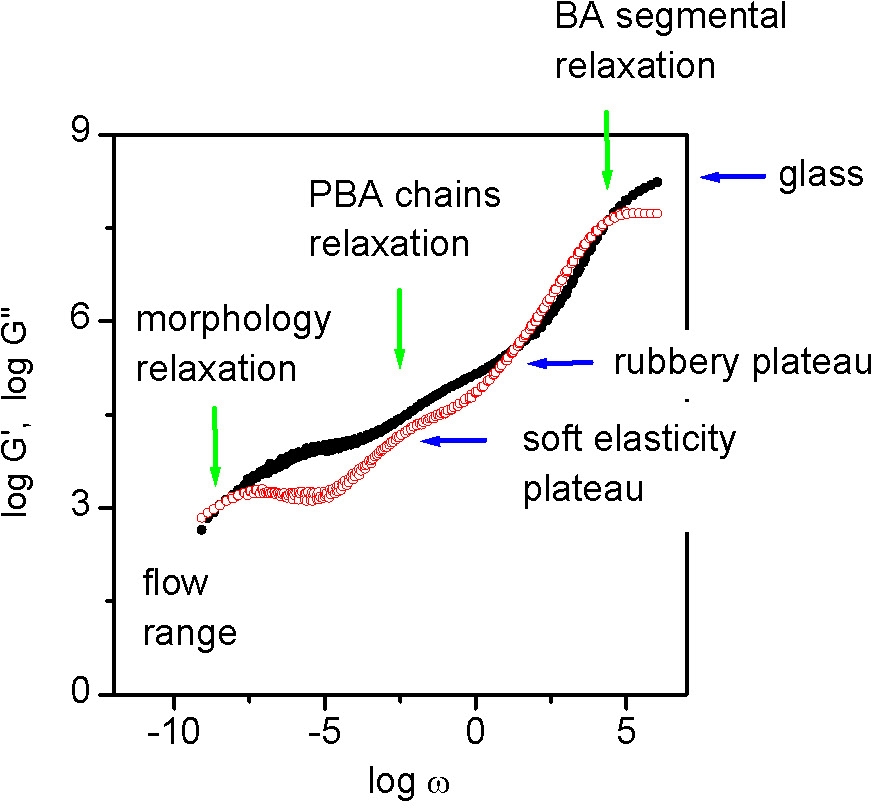
Changes in the copolymer architecture influence the range of frequencies (or temperatures) in which various properties appear. The fabricated material behaves in the manner expected from a pBA with a molecular weight in the millions due to the requirement for molecular relaxation.
This requirement for cooperative molecular relaxation and the consequential affect on physical properties is perhaps more dramatically illustrated by an examination of the relaxation modes of a melt of bottle-brush macromolecules. When the relaxation modes of a poly(n-butylacrylate) bottle-brush macromolecules with a main chain length of 400 and a side chain length of 60 is examined one can discern an additional relaxation resulting from the total molecular relaxation at very low modulus. Because of the spatial correlations of the supramolecular units and the dense packing of polymer chains in bulk materials the motion must occur cooperatively and therefore constitutes the slowest structural relaxation.
This phenomenon is also seen in single bottle brush macromolecules which display comparable complexity and indeed were the first materials in which the supersoft state was identified.(3)
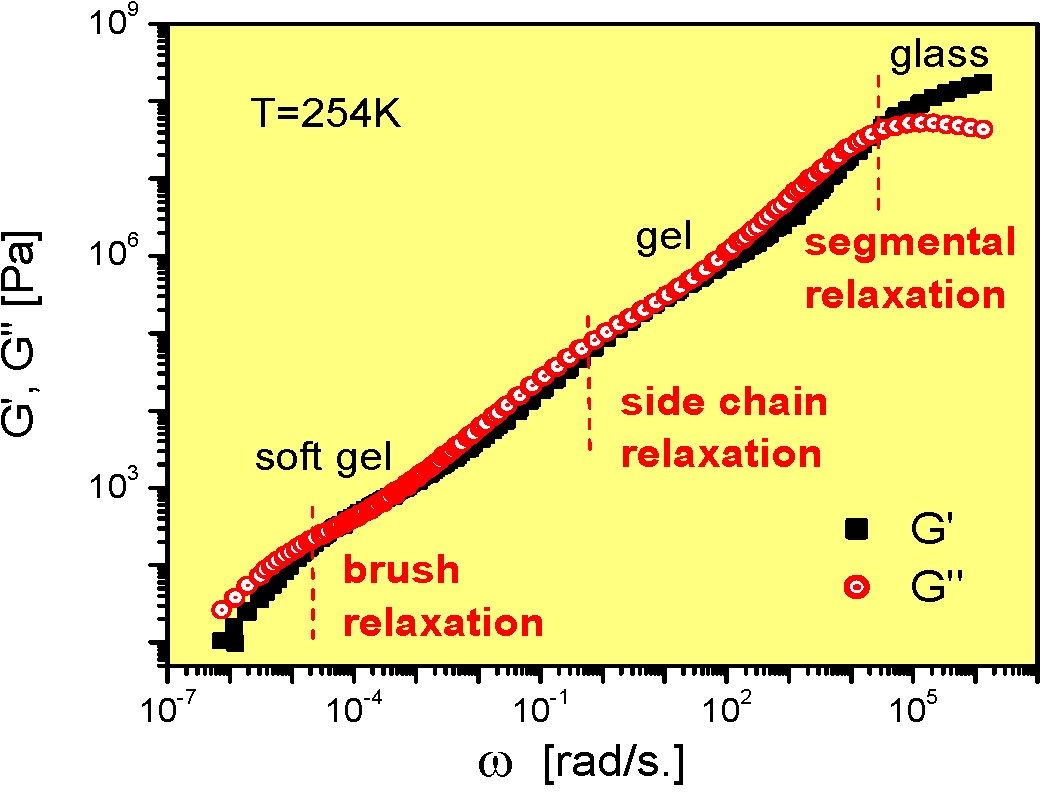
The side chain length and backbone length can be independently varied and each influences the viscoelastic behavior of the bottlebrush copolymers. The backbone length has a major influence on the flow controlled by global rearrangements. The properties are discussed in detail below.
Another aspect of molecular assembly of block copolymers is discussed in the section on precursors for carbon nano-structures where phase separated block copolymers containing PAN are converted to carbon nanostructures.
Controlling morphology by PDI:
A new phenomenon was observed when examining the morphology of polystyrene-b-poly(metyl acrylate) (PS-PMA) copolymers that had been synthesized using activators regenerated by electron transfer (ARGET) for atom transfer radical polymerization (ATRP). It was observed that the polydispersity of the PMA block could be varied by adjusting the amount of copper catalyst in ARGET ATRP. The resulting broader molecular weight distributions were found to be approximately symmetrical. A hexagonally perforated lamellar (HPL) morphology was observed for a polydisperse PS-PMA copolymer when the block copolymer contained ~35 vol % of PMA after short- and long-term solvent-casting conditions.(4)
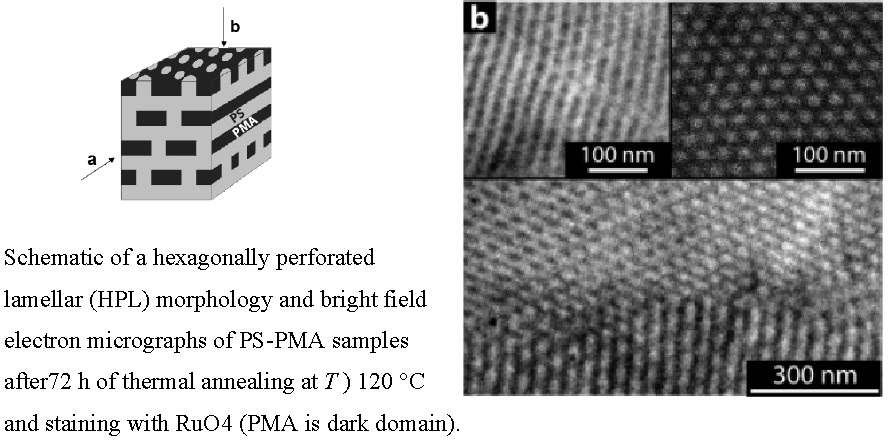
No order-order transitions were observed at elevated temperature or after prolonged thermal annealing. The stabilization of the HPL morphology, a morphology that is considered to be metastable in narrow-disperse diblock copolymers as well as diblock copolymers with selective block polydispersity given by a Schulz-Zimm distribution-suggests that the skewness of the distribution of block molecular weight is an important parameter for the structure selection during the microphase separation process. In particular, the results indicate that near-symetrical block molecular weight distributions (as realized by the ARGET ATRP technique) facilitate the stabilization of microdomain morphologies with increased standard deviation of mean curvature. The results point to the relevance of controlling both the width and symmetry of molecular weight distribution as a potential route toward the tailored synthesis of nonregular microstructures with particular topological properties that might be of future technological interest such as membranes.
Multisegmented blocks:
Multisegmented block copolymers were prepd. by the step-growth click coupling of well- defined block copolymers synthesized by ATRP).(5) In the following schematic a low molecular weight polystyrene was prepared from a difunctional initiators, Br-PS-Br then the bromo end groups were transformed to azido groups by simple nucleophilic substitution, and the resulting N3-PS-N3 was coupled by reaction with Pg2O in DMF with a CuBr catalyst.(6)
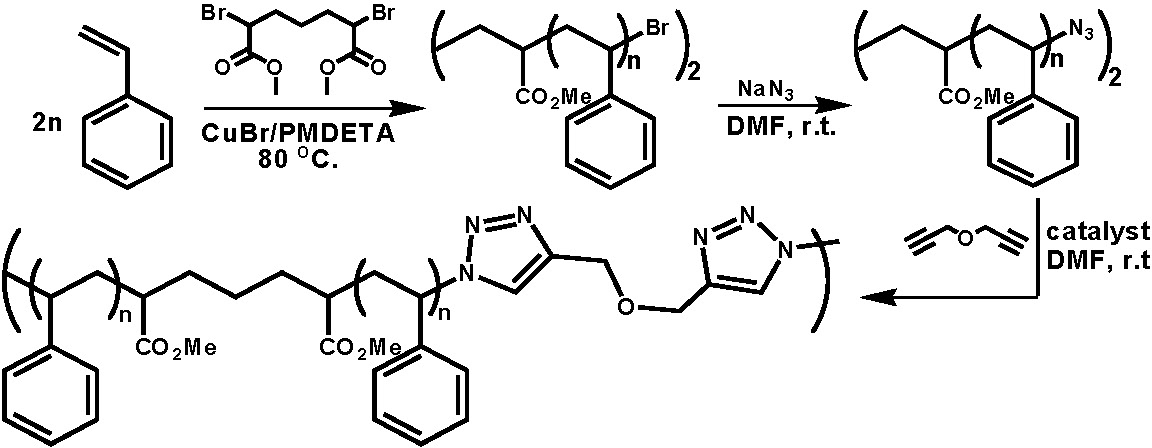
In further examples amphiphylic multisegmented block copolymers were prepared from a diazido-terminated polystyrene-b-poly(ethylene oxide)-b-polystyrene ABA block copolymer which was coupled with propargyl ether in N,N-dimethylformamide in the presence of a CuBr/PMDETA catalyst.(7) The preparation of multisegmented block copolymers was also demonstrated by the click coupling of propargyl ether with another diazido-terminated triblock copolymer, poly(butyl acrylate)-b-poly(methyl methacrylate)-b-poly(butyl acrylate), and a diazido-terminated pentablock copolymer; polystyrene-b-poly(butyl acrylate)-b-poly(methyl methacrylate)-b-poly(butyl acrylate)-b-polystyrene.
The formation of a product of higher MW and broader MWD was verified by triple-detection size exclusion chromatography, which revealed that typically five to seven block copolymers were linked together during the click reaction. Differential scanning calorimetry and dynamic mechanical analysis revealed that the amphiphilic block copolymer behaves as a viscoelastic fluid, while its corresponding multiblock copolymer is an elastic material. The multisegmented block copolymers with partially miscible segments exhibit higher glass transition temperatures than their precursors.(7)
Degradable multiblock copolymers were also prepared by a different type of click coupling chemistry. In this case the multi-block copolymers were prepared by radical cross-coupling of α,ω-dihalogenated polymers prepared by ATRP with disulfide containing dinitroxides. High MW polymers (Mn > 100,000 g mol-1) with more than 10 cleavable units in the main chain were prepared. These multisegmented polymers were chemically and thermally degraded. Chemical degradation via basic hydrolysis or disulfide reduction, as well as thermal degradation by heating in the presence or in the absence of free nitroxides, led to low MW polymers with relatively low polydispersity.(8)
REFERENCES
(1) Gaynor, S. G.; Matyjaszewski, K. Macromolecules 1997, 30, 4241-4243.
(2) Zhang, Y.; Chung, I. S.; Huang, J.; Matyjaszewski, K.; Pakula, T. Macromolecular Chemistry and Physics 2005, 206, 33-42.
(3) Pakula, T.; Matyjaszewski, K. In PCT Int. Appl.; (Carnegie Mellon University, USA). WO 2004014963, 2004; p 65 pp.
(4) Listak, J.; Jakubowski, W.; Mueller, L.; Plichta, A.; Matyjaszewski, K.; Bockstaller, M. R. Macromolecules 2008, 41, 5919-5927.
(5) Golas, P. L.; Matyjaszewski, K. QSAR & Combinatorial Science 2007, 26, 1116-1134.
(6) Golas, P. L.; Tsarevsky, N. V.; Sumerlin, B. S.; Matyjaszewski, K. Macromolecules 2006, 39, 6451-6457.
(7) Golas, P. L.; Tsarevsky, N. V.; Sumerlin, B. S.; Walker, L. M.; Matyjaszewski, K. Australian Journal of Chemistry 2007, 60, 400-404.
(8) Nicolaye, R.; Marx, L.; Hemery, P.; Matyjaszewski, K. Macromolecules 2007, 40, 9217-9223.