ARGET ATRP and ICAR ATRP
Activator ReGenerated by Electron Transfer (ARGET) ATRP
In many ways, Activators ReGenerated by Electron Transfer (ARGET) is not just another way to initiate an ATRP but is a new way to run a CRP; a “green” procedure that uses much lower concentrations of catalysts in a system that is suited for industrial scale up, and produces pure α-functional products.
When we considered the implications of the convenient AGET procedure for initiating an ATRP, described above, where the activators are generated by electron transfer, we realized that it should be possible to use the reducing agents to constantly regenerate the ATRP activator, the CuI species, from CuII species irreversibly formed during termination processes, without directly or indirectly producing initiating species that generate new chains.(1) The amount of Cu-based catalysts in atom transfer radical polymerization (ATRP) of styrene could therefore be reduced to a few ppm in the presence of the appropriate reducing agents such as FDA approved tin(II) 2-ethylhexanoate (Sn(EH)2), glucose,(1, 2) ascorbic acid, (3) silver metal,(4, 5) hydrazine or phenyl hydrazine.(6) The rate of reduction, and hence the rate of polymerization, can be adjusted by the type and amount of reducing agent or by slow/intermittent feeding of the reducing agent if one desires to reduce the initial concentration of CuI-based catalyst in a very active polymerization system.(7, 8) Selection of a suitable reducing agent is of great importance, since one needs to ensure that both reducing agent, and its oxidized product, does not interfere with any of the reagents added for the desired ATRP.(9) Examples of side reactions include complexation or reaction with the ligand, monomer, or polymer, complexation of reducing agent with the metal center of the catalyst complex, protonation of the ligand by acidic oxidation products, or nucleophilic substitution or elimination of halide chain ends.(10, 11) These side reactions can lead to poor control. In a specific example, SnII-mediated ARGET ATRP technique was found to be unsuitable reducing agent for the polymerization of glycidyl methacrylate as it resulted in polymers with multimodal molecular weight distributions due to the occurrence of epoxide ring-opening reactions, catalyzed by SnII and SnIV. (9) Nevertheless, appropriately selected reducing agents allow starting an ATRP with the oxidatively stable CuII species and the reducing/reactivating cycle can be employed to eliminate air or radical traps in the system with only ppm levels of added catalyst. For example, styrene was polymerized by the addition of 5 ppm of CuCl2/Me6TREN and 500 ppm of Sn(EH)2 to the reaction resulting in preparation of a polystyrene with Mn = 12,500 (Mn,th = 12,600) and Mw/Mn = 1.28.(12)
ARGET ATRP has also been applied to polymerization from surfaces even in the presence of limited amounts of air.(13)
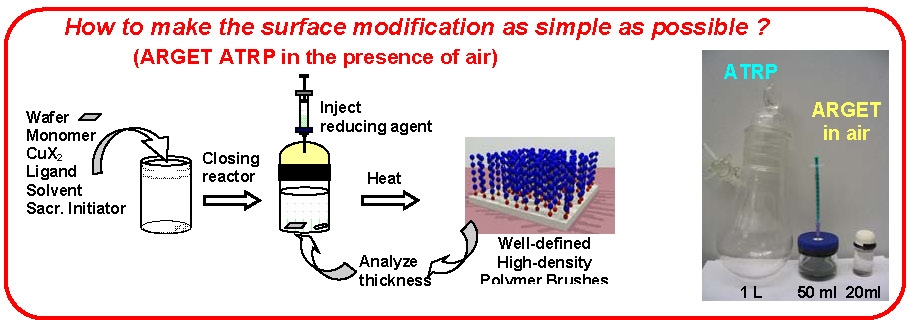
Generally, in an ARGET system it is desirable to add an excess of the ligand compared to the amount required to form the transition metal complex in order to compensate for competitive complexation of the low amount of added transition metal with monomer/solvent/reducing agent that are all present in significant molar excess. Indeed it has been determined that the ARGET procedure can be driven based solely on addition of excess ligand, or ligand substitute,(14, 15) and more recently by a nitrogen containing monomer, 2-(dimethylamino) ethyl methacrylate (DMAEMA).(16) This indicated that nitrogen containing compounds such as DMAEMA and triethylamine are actually acting more as reducing agents than ligand substitutes.(17)
Another advantage of ARGET ATRP is that copper catalyst induced side reactions, particularly for acrylates,(18-20) are also reduced to a significant degree and it is now possible to drive an ATRP reaction to much higher conversion and prepare copolymers with much higher molecular weight (13, 21) while retaining chain end functionality. This has been confirmed by successful chain extension of macromolecules formed using this initiation/continuous reactivation system.(22) Catalyst selection is important in an ARGET ATRP procedure so that a controlled polymerization is attained and an expedited procedure for catalyst optimization has been examined.(10, 23) ARGET ATRP was successfully extended to aqueous media(8) which allowed preparation of protein-polymer hybrids in a grafting from reaction(24) and development of photoinduced ATRP with ppm-levels of Cu catalyst by visible light in aqueous media.(25) Molecular bottlebrushes(26) as well as composites (26-28) have been prepared with ppm amounts of Cu catalyst.(29)
Initiators for Continuous Activator Regeneration (ICAR) ATRP
The concept of Initiators for Continuous Activator Regeneration (ICAR) could simplistically be considered a “reverse” ARGET ATRP[p1] as a source of free radicals is employed to slowly and continuously regenerate the very low concentration of CuI activator which is otherwise consumed in termination reactions in ICAR ATRP. The rate of decomposition of the selected free radical initiator should be slow and a fraction of the initiator should remain at the end of the reaction, otherwise the reaction would stop. This means that the kinetics of an ICAR ATRP resembles a standard FRP or a RAFT polymerization and depends on the rate of decomposition of the added initiator and not on the ATRP equilibrium constants.(7, 30) ICAR ATRP is similar to SR&NI ATRP or RAFT in that a small fraction of chains can originate from the added radical initiator. Slow feeding of the added initiator can be used to mediate the rate of polymerization and avoid/reduce the possibility of poor temperature control.(31) Nevertheless, with this technique, controlled synthesis of polystyrene and poly(meth)acrylates (Mw/Mn < 1.2) can be conducted with catalyst concentrations between 10-50 ppm, where removal or recycling of the catalyst complex would be unwarranted for many applications. The reaction is driven to completion with low concentrations of added standard free radical initiators,(21) providing a procedure that could be conducted in existing industrial polymerization equipment.
Kinetic modeling was used to better understand and optimize initiators for continuous activator regeneration atom transfer radical polymerization (ICAR ATRP). The polymerization conditions were adjusted as a function of the ATRP catalyst reactivity for two monomers, methyl methacrylate and styrene. In order to conduct a well-controlled ICAR ATRP process with a low amount of catalyst (ppm level), a sufficiently low initial concentration of conventional radical initiator relative to the initial ATRP initiator is required. In some cases, stepwise addition of a conventional radical initiator is needed to reach high conversion. Under such conditions, the equilibrium of the activation/deactivation process for macromolecular species can be established at low conversion.(32)
Four ATRP catalysts with a broad range of KATRP values were examined in ICAR ATRP of styrene. They included the CuCl2 complexes with tris[2-(dimethylamino)ethyl]amine (Me6TREN), tris[(2-pyridyl)methyl]amine (TPMA) as ligands, both available from Maruzen Chemical, N,N,N’,N”,N”-pentamethyldiethylenetriamine (PMDETA), and 4,4'-di-(5-nonyl)-2,2'-bipyridine (dNbpy). ICAR ATRP of styrene was first conducted at low temperature (60°C) where organic radicals were produced solely by the slow decomposition of azobisisobutyronitrile (AIBN) (0.1 eq vs. ethyl 2-bromoisobutyrate (EtBrIB) initiator) in the presence of 50 ppm of CuCl2/L complexes. Interestingly, rates of polymerization differed by less than a factor of two amongst the reactions conducted with the different catalyst complexes. This was initially surprising, given that values of KATRP, which govern radical concentration and the rate of polymerization under normal and SR&NI ATRP conditions, differ by more than four orders of magnitude among these four complexes. Additional experiments and kinetic simulations explored the possibility that: (a) rates of polymerization and radical concentration under ICAR ATRP conditions are actually controlled by the rate of free radical initiator decomposition and, (b) the relative CuI and CuII concentrations conform accordingly as dictated by the KATRP value. Since a very small amount of Cu catalyst is employed in ICAR ATRP, catalysts with large values of KATRP (high concentration of CuII) and fast deactivation rate constants will minimize this ratio, allowing for a more even polymer chain growth and ultimately better control.
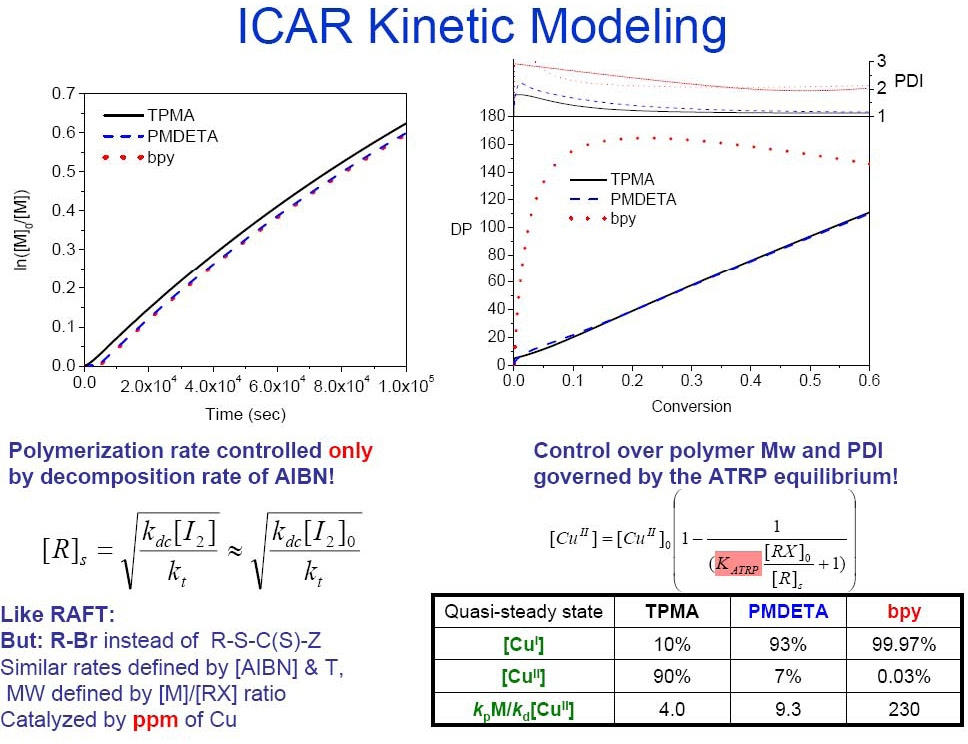
Cu complexes with TPMA have a high value for KATRP with the model polystyrene chain end 1-(bromoethyl)benzene (~7.9×10-6 at 60oC). While the analogous KATRP value of the Cu/PMDETA complex is much lower (~ 5.9×10-8), the deactivation rate constant (kda) for Cu/PMDETA is larger than that of TPMA, which can compensate for the product of kda[CuII]. Therefore Me6TREN and TPMA are more suitable ligands than PMDETA and dNbpy for ICAR ATRP at low Cu catalyst concentrations.
Simulations confirmed that the rate of polymerization in ICAR is governed by the rate of free radical initiator decomposition (as in RAFT) while control is ultimately determined by KATRP and the rate of deactivation (as in ATRP).(22) ICAR ATRP was also extended to iron-based catalysts and a successful polymerization of styrene was carried out with ppm amounts of FeIIIBr3 and 1,1'-azobis(cyclohexanecarbonitrile)(33) and for ICAR of MMA with 50 ppm iron-based N-heterocyclic carbine, (FeBr3(NHC)), complexes in 50% vol./vol. anisole resulted in PMMA providing after 24 h at T = 60 °C a polymer with Mw/Mn = 1.20 at 65% monomer conversion.(34) ICAR was also successful for preparation of styrene and MMA with Ru(Cp*)Cl(PPh3)2.(35) ICAR ATRP with ppm Cu Catalyst was also successful in water.(36)
AGET ATRP, ARGET ATRP and ICAR ATRP are beginning to gain increasing attention because of the ease of setting up the reaction and the low amounts of catalyst employed in the reaction. (16, 37-40) Furthermore ICAR ATRP can be conducted in existing industrial equipment using existing amounts of free radical initiators resulting in controlled radical polymerization being conducted with a similar rate of conversion to current uncontrolled polymerizations.
Borderline between SR&NI ATRP and ICAR ATRP
A recent article noted SR&NI ATRP and ICAR ATRP are phenomenologically very similar. In both methods, thermal radical initiators are employed to reduce the catalyst in the higher oxidation state and generate CuI activator in situ and set out to determine if there was a boundary between the procedures.(41) The article is of particular interest as it describes parameters for selection of the correct catalysts for each procedure. A similar analysis is relevant for AGET ATRP and ARGET ATRP but was not carried out as the borderline between AGET and ARGET systems is challenging due to a more complex reduction kinetics and side reactions such as the possible ligand displacement from the Cu complex by tin(II)octoate, a commonly used reducing agent. Nevertheless, the borderline criterion should be similar. Thus the reduction rate of the reducing agent should be known in order to distinguish between the two mechanisms. A commonly used chemical reducing agent, ascorbic acid, can be used in both AGET and ARGET ATRP.
SR&NI and ICAR ATRP generally differ in the amount of catalyst used and in the rate of catalyst reduction. Both procedures have been extensively studied and were the subject of detailed kinetic investigations. (32, 42, 43) Commonly, SR&NI ATRP requires higher catalyst loadings and a quick initial reduction of CuII, while ICAR ATRP relies on slow and continuous reduction of smaller catalyst loadings. The article investigated both methods through kinetic simulations and established a borderline kinetic criterion. If the polymerization rate depends on the rate of decomposition of the radical initiator, the system follows ICAR ATRP kinetics, and if it depends on the ATRP equilibrium constant, it follows SR&NI ATRP. PREDICI, which has been shown accurately simulate radical polymerization procedures,(7, 30, 44-46) was employed to determine the differences. PREDICI simulations were performed for two monomers, methyl methacrylate (MMA) and a hypothetical acrylate monomer with the kinetic properties of methyl acrylate, which however does not undergo backbiting reactions (MA*). The transition from one to the other mechanism occurred continuously with an inflection point at a ratio of rate coefficients of radical initiator decomposition to propagation of about kdc/kp ≈ 10−7 m under typical conditions. For faster initiator decomposition and slower propagation, ATRP follows SR&NI ATRP and for slower decomposition and faster propagation it obeys ICAR ATRP kinetics.
REFERENCES
(1) Jakubowski, W.; Min, K.; Matyjaszewski, K. Macromolecules 2006, 39, 39-45.
(2) Jakubowski, W.; Matyjaszewski, K. Angew. Chem., Int. Ed. 2006, 45, 4482-4486.
(3) Min, K.; Gao, H.; Matyjaszewski, K. Macromolecules (Washington, DC, United States) 2007, 40, 1789-1791.
(4) Williams, V. A.; Matyjaszewski, K. Macromolecules 2015, 48, 6457-6464.
(5) Williams Valerie, A.; Ribelli Thomas, G.; Chmielarz, P.; Park, S.; Matyjaszewski, K. J Am Chem Soc 2015, 137, 1428-1431.
(6) Matyjaszewski, K.; Jakubowski, W.; Min, K.; Tang, W.; Huang, J.; Braunecker, W. A.; Tsarevsky, N. V. Proceedings of the National Academy of Sciences of the United States of America 2006, 103, 15309-15314.
(7) Krys, P.; Ribelli, T. G.; Matyjaszewski, K.; Gennaro, A. Macromolecules 2016, 49, 2467-2476.
(8) Simakova, A.; Averick, S. E.; Konkolewicz, D.; Matyjaszewski, K. Macromolecules 2012, 45, 6371-6379.
(9) Tsarevsky, N. V.; Jakubowski, W. Journal of Polymer Science, Part A: Polymer Chemistry 2011, 49, 918-925.
(10) Woodruff, S. R.; Davis, B. J.; Tsarevsky, N. V. ACS Symposium Series 2012, 1100, 99-113.
(11) Matyjaszewski, K.; Tsarevsky, N. V. Journal of the American Chemical Society 2014, 136, 6513-6533.
(12) Matyjaszewski, K.; Dong, H.; Jakubowski, W.; Pietrasik, J.; Kusumo, A. Langmuir 2007, 23, 4528-4531.
(13) Pietrasik, J.; Dong, H.; Matyjaszewski, K. Macromolecules 2006, 39, 6384-6390.
(14) Matyjaszewski, K.; Jakubowski, W.; Spanswick, J. In WIPO; (Carnegie Mellon University, USA). Application: WO 2007025310, 2007; p 95pp.
(15) Kwak, Y.; Matyjaszewski, K. Polymer International 2009, 58, 242-247.
(16) Dong, H.; Matyjaszewski, K. Macromolecules 2008, 41, 6868-6870.
(17) Tang, H.; Shen, Y.; Li, B.-G.; Radosz, M. Macromolecular Rapid Communications 2008, 29, 1834-1838.
(18) Schroder, K.; Konkolewicz, D.; Poli, R.; Matyjaszewski, K. Organometallics 2012, 31, 7994-7999.
(19) Matyjaszewski, K.; Woodworth, B. E. Macromolecules 1998, 31, 4718-4723.
(20) Ribelli, T. G.; Wahidur Rahaman, S. M.; Krys, P.; Matyjaszewski, K.; Poli, R. Macromolecules 2016, 49, 7749-7757.
(21) Dong, H.; Tang, W.; Matyjaszewski, K. Macromolecules 2007, 40, 2974-2977.
(22) Mueller, L.; Jakubowski, W.; Tang, W.; Matyjaszewski, K. Macromolecules 2007, 40, 6464-6472.
(23) Siegwart, D. J.; Leiendecker, M.; Langer, R.; Anderson, D. G. Macromolecules 2012, 45, 1254-1261.
(24) Averick, S. E.; Bazewicz, C. G.; Woodman, B. F.; Simakova, A.; Mehl, R. A.; Matyjaszewski, K. European Polymer Journal 2013, 49, 2919-2924.
(25) Pan, X.; Malhotra, N.; Simakova, A.; Wang, Z.; Konkolewicz, D.; Matyjaszewski, K. Journal of the American Chemical Society 2015, 137, 15430-15433.
(26) Song, Y.; Ye, G.; Lu, Y.; Chen, J.; Wang, J.; Matyjaszewski, K. ACS Macro Letters 2016, 5, 382-386.
(27) Iacono, M.; Heise, A. Polymers (Basel, Switzerland) 2015, 7, 1427-1443.
(28) Askar, S.; Li, L.; Torkelson, J. M. Macromolecules 2017, Ahead of Print.
(29) Nese, A.; Li, Y.; Sheiko, S. S.; Matyjaszewski, K. ACS Macro Letters 2012, 1, 991-994.
(30) Zhong, M.; Matyjaszewski, K. Macromolecules 2011, 44, 2668-2677.
(31) Jakubowski, W. ACS Symposium Series 2012, 1100, 203-216.
(32) D'Hooge, D. R.; Konkolewicz, D.; Reyniers, M.-F.; Marin, G. B.; Matyjaszewski, K. Macromol. Theory Simul. 2012, 21, 52–69,.
(33) Mukumoto, K.; Wang, Y.; Matyjaszewski, K. ACS Macro Letters 2012, 1, 599-602.
(34) Okada, S.; Park, S.; Matyjaszewski, K. ACS Macro Letters 2014, 3, 944-947.
(35) Plichta, A.; Li, W.; Matyjaszewski, K. Macromolecules 2009, 42, 2330-2332.
(36) Konkolewicz, D.; Magenau, A. J. D.; Averick, S. E.; Simakova, A.; He, H.; Matyjaszewski, K. Macromolecules 2012, 45, 4461-4468.
(37) Tanaka, K.; Matyjaszewski, K. Macromolecular Symposia 2008, 261, 1-9.
(38) Chan, N.; Cunningham, M. F.; Hutchinson, R. A. Macromolecular Chemistry and Physics 2008, 209, 1797-1805.
(39) Pintauer, T.; Matyjaszewski, K. Chemical Society Reviews 2008, 37, 1087-1097.
(40) Yamamoto, S.-i.; Matyjaszewski, K. Polymer Journal (Tokyo, Japan) 2008, 40, 496-497.
(41) Krys, P.; Schroeder, H.; Buback, J.; Buback, M.; Matyjaszewski, K. Macromolecules 2016, 49, 7793-7803.
(42) Tang, W.; Matyjaszewski, K. Macromolecular Theory and Simulations 2008, 17, 359-375.
(43) Toloza Porras, C.; D'Hooge, D. R.; Reyniers, M.-F.; Marin, G. B. Macromolecular Theory and Simulations 2013, 22, 136-149.
(44) Soerensen, N.; Schroeder, H.; Buback, M. Macromolecules (Washington, DC, United States) 2016, 49, 4732-4738.
(45) Schroeder, H.; Buback, J.; Schrooten, J.; Buback, M.; Matyjaszewski, K. Macromolecular Theory and Simulations 2014, 23, 279-287.
(46) Schroeder, H.; Buback, M. Macromolecules (Washington, DC, United States) 2014, 47, 6645-6651.