Atom Transfer Radical Polymerization (ATRP)
ATRP is mechanistically related to transition metal mediated atom transfer radical addition (ATRA) reactions(1) and indeed this relationship was the reason this transition metal mediated controlled radical polymerization process was named ATRP.(2) ATRP can be viewed as a very special case of ATRA. However, in contrast to ATRA reactions, ATRP requires reactivation of the first formed alkyl halide adduct with the unsaturated compound (monomer) and the further reaction of the intermittently formed radical with additional monomer units (propagation). The "livingness" of this polymerization process can be ascertained from a linear first-order kinetic plot, accompanied by a linear increase in polymer molecular weight with conversion, with the value of the number-average degree of polymerization (DPn) determined by the ratio of reacted monomer to initially introduced initiator (i.e., DPn = D[M]/[RX]0).
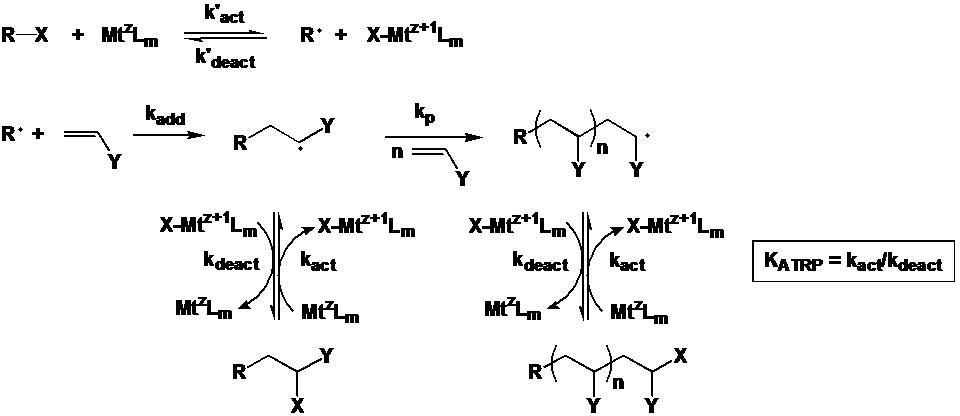
Scheme 1.Mechanism of metal complex-mediated ATRA and ATRP.(3)
The normal schematic of the ATRP equilibrium, which emphasizes the repetitive nature of the activation and deactivation steps and the need to push the equilibrium to the left hand side, thereby forming a low concentration of radicals to reduce radical-radical termination reactions, and ensure a high mole fraction of dormant chains, is shown below.
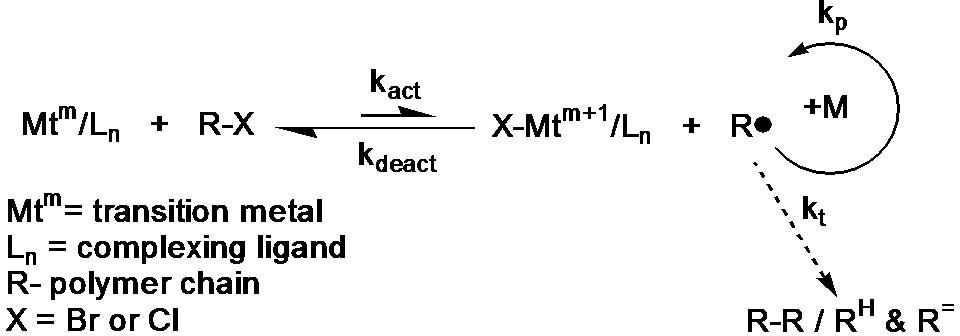
Scheme 2. Mechanism of metal complex-mediated ATRP
The “livingness” of this polymerization process can be ascertained from a linear first-order kinetic plot, accompanied by a linear increase in polymer molecular weight with conversion, with the value of the number-average degree of polymerization (DPn) determined by the ratio of reacted monomer to initially introduced initiator (i.e., DPn = D[M]/[RX]0).
ATRP is based on an inner sphere electron transfer process,(4) which involves a reversible homolytic (pseudo)halogen transfer between a dormant species, an added initiator or dormant propagating chain end (R-X or R-Pn-X) and a transition metal complex in the lower oxidation state (Mtm/Ln), resulting in the formation of propagating radicals (R●)(5) and the metal complex in the higher oxidation state with a coordinated halide ligand (e.g. X-Mtm+1/Ln). The active radicals form with a measurable rate constant of activation kact, subsequently propagate with a rate constant kp and are reversibly deactivated kdeact, but, since ATRP is a radical based process, the active species can also terminate with a rate constant kt. As the reaction progresses radical termination is diminished as a result of the persistent radical effect (PRE),(6,7) increased chain length, as well as conversion and viscosity.(8) Consequently, the equilibrium is strongly shifted towards the dormant species (kact << kdeact).(7)
There are four main variables that influence KATRP; temperature,(9-10) pressure,(11-12) media/solvent (which increases with polarity: toluene <anisole <MeCN <DMF <DMSO <H2O)(13) and obviously alkyl halide and catalyst.(14) The higher oxidation state transition metal (complex), the equivalent of the persistent radical in an ATRP, can be added directly to a reaction prior to initiation to increase the efficiency of initiation, by reducing the fraction of low molecular weight termination reactions initially required to generate the PRE,(15) or can be formed in situ by reaction with dissolved oxygen.(16) Addition of the persistent radical, (Mtm+1 in the case of ATRP) is of particular utility when conducting a “grafting from” reaction with a multifunctional initiator or grafting from a surface. It is also strongly recommended when ATRP is carried out in protic, particularly homogeneous aqueous media.(17)
ATRP is in many ways a complex reaction, since the polymerization process includes one or more (co)monomers, a transition metal complex in two or more oxidation states,(18) which can comprise various counterions and ligands, an initiator with one or more radically transferable atoms or groups and an optional solvent, suspending media or various additives. All of the components present in the reaction medium can, and often do, affect the interactions between the reagents that constitute the ATRP equilibrium.(19,20) The initiator is most frequently an alkyl (pseudo)halide which can be either a low or high molar mass compound or even a part of an insoluble material, such as when initiators are tethered to the surface of modified particles, flat wafers, or even fibers, etc.
A series of starting points for conducting an ATRP for a range on monomers in different media is provided elsewhere on this site.
In most of the following discussion we will use copper(21) as an exemplary transition metal but a wide range of other metals can be employed in an ATRP including Ti,(22) Mo,(23-25) Re,(26-27) Fe,(28-34) Ru,(35-38) Os,(39-40) Rh,(41-42) Co,(43) Ni,(44-47) and Pd.(48) There are additional references in a recent review article which also address the environmental issues associated with industrial scale ATRP,(49) indeed from an environmental and cost perspective recent work on iron based catalysts provide an incentive for further examination of such systems.(34),(50-51) In one paper ATRP of MMA in polar solvents, such as NMP, DMF and MeCN, was successfully catalyzed by FeBr2 in the absence of any additional ligand. The solvents probably play the role of efficient ligands for FeBr2. NMP provided the highest activity among the solvents investigated for FeBr2 catalyzed ATRP of MMA. Molecular weights agreed well with theoretical values and molecular weight distributions were relatively narrow (Mw/Mn=1.26~1.28). Addition of a small amount of FeBr3 improved the level of control in these systems. This system provides a low cost and environmentally friendly ATRP process.Nevertheless copper has proven by far to be the transition metal of choice, as determined by the successful application of a spectrum of copper complexes as catalysts, for the ATRP of a broad range of monomers in diverse media by many research groups. However iron may eventually prove to be the transition metal of choice for environmental reasons unless industrially viable procedures for internal reuse of the copper complexes are adopted. It should also be noted that Ru and Os have certain advantages as a consequence of their high halidophilicity that may make them a good choice for use in protic media.(40)
Polymers prepared by other polymerization processes can be functionalized at the termini or along the backbone and incorporated into an ATRP as a macromonomer or macroinitiator, or simultaneously through use of both a macroinitiator and a macromonomer to improve incorporation of the macromonomer into the polymer,(52) leading to the preparation of well defined block and graft copolymers. There may be multiple initiating sites in either a small molecule or a macroinitiator, leading to chain growth in several directions. A functional initiator may carry a second non-initiating functionality, in addition to a radically transferable atom or group, to yield telechelic materials.(53)
The transition metal complex has to be at least partially soluble in the reaction medium and reactions can be run under homogeneous or heterogeneous conditions. The former generally provides better control since the concentration of activator and deactivator can be controlled.(54) Reaction temperatures typically range from room temperature to 150 0C, but can be correspondingly altered. The reaction can be run under vacuum or pressure.(55) Reactions can not only be conducted in the presence of moisture but even in the presence of water under homogeneous(56) or heterogeneous (micro-emulsion → suspension) conditions.(57-58) Oxygen should be removed from the reaction medium, but a limited amount of oxygen can be tolerated particularly in the presence of an added reducing agent e.g. Cu(0), Sn(EH2), ascorbic acid, reducing sugars or amines.(18,59-64) The order of addition of reagents may vary but most often the initiator, or catalyst activator, is the last reagent to be added to a preformed solution of the catalyst in the monomer/solvent. An important parameter may be the addition or formation of a small amount of the Cu(II) species at the beginning of the reaction, since it enables the deactivation process to occur immediately without requiring its spontaneous formation by termination reactions, thereby providing both higher initiator efficiency, reduced cost and instantaneous control.(65)
Understanding, and controlling the equilibrium, and hence the dynamics of the atom transfer process, are basic prerequisites for running a successful ATRP. Therefore it remains a very important objective within the Matyjaszewski group to correlate structure with reactivity for each of the involved reagents, the oxidation states of the transition metal complex, radicals and dormant species, in addition to solvent effects and reaction temperature in order to provide a fundamental understanding of the reaction kinetics required for the selection of optimum conditions to conduct the desired reaction.(14,18,66-71)
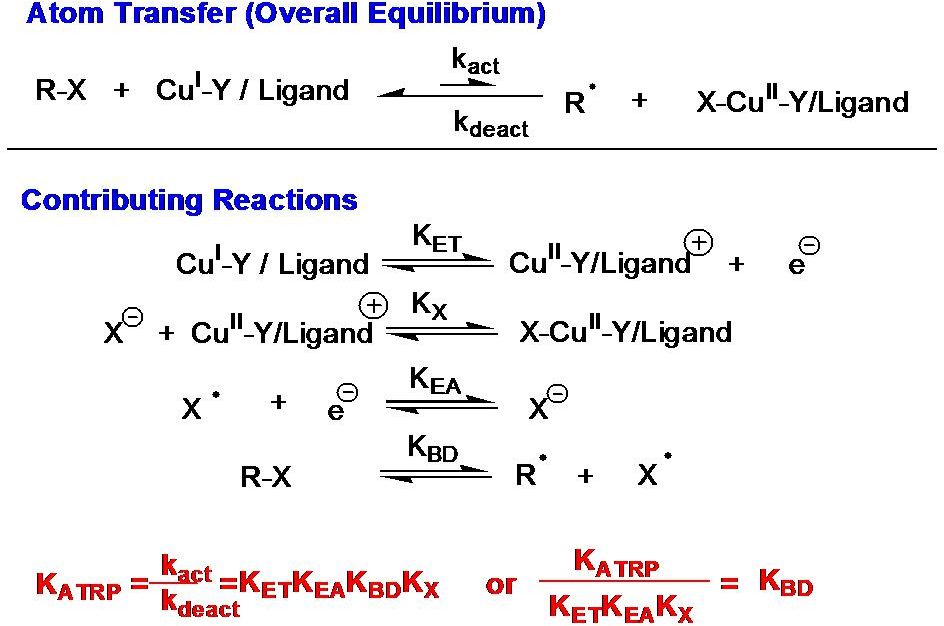
The ATRP equilibrium, (KATRP in the scheme above), is expressed as a combination of several contributing reversible reactions, including a combination of C-X bond homolysis of the alkyl halide (R-X), (other transferable atoms or groups can participate in an ATRP(72) but the most frequently employed radically transferable atoms are halogens and will be used to exemplify ATRP throughout most of following discussions) two redox processes, and heterolytic cleavage of the CuII-X bond. Therefore KATRP can be expressed as the product of the equilibrium constants for electron transfer between metal complexes (KET), electron affinity of the halogen (KEA), bond dissociation energy of the alkyl halide (KBD) and the equilibrium constant for the heterolytic cleavage of the Mtn+1-X bond (KX), which measures the “halidophilicity” of the deactivator. This means that for a given alkyl halide, R-X, the activity of a catalyst in an ATRP reaction not only depends on the redox potential, but also on the halidophilicity of the transition metal complex. For complexes that have similar halidophilicity, the redox potential can be used as a measure of catalyst activity in the ATRP. This was demonstrated by the linear correlation between KATRP and E1/2, (ie. KET), for a series of CuI complexes with nitrogen based ligands.(70,73-74)
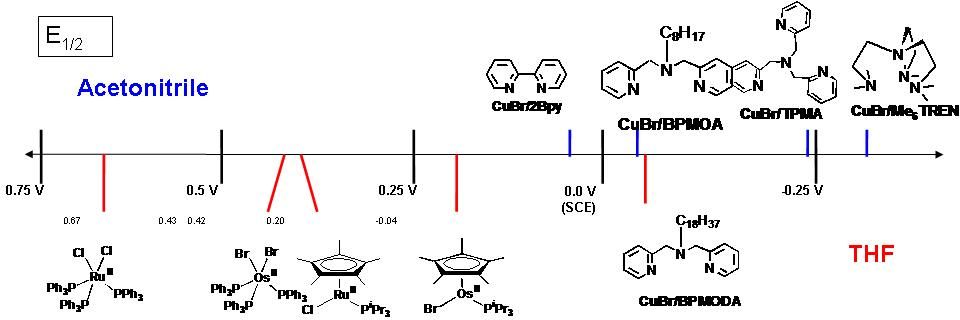
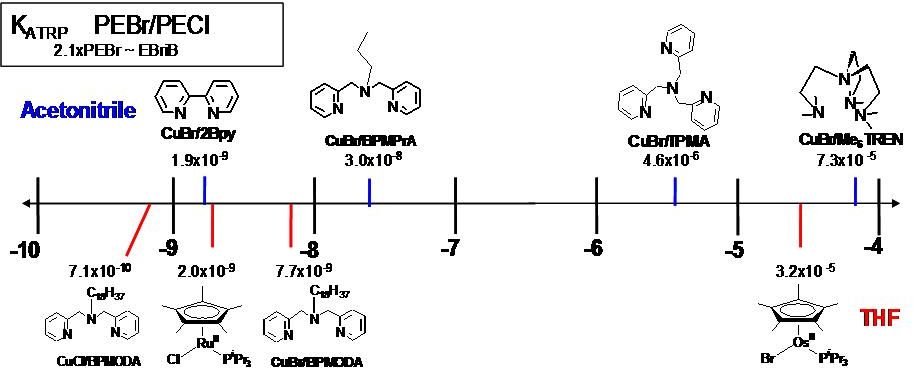
It can also be concluded that for a given catalytic system in the same solvent (where KET, KEA, and KX are essentially constant), KATRP should depend upon the energy required for alkyl halide bond homolysis, or KBD. Indeed, when the alkyl halide bond dissociation energies were calculated for a series of ATRP monomers/initiators, they were found to correlate well with measured values of KATRP.(68,73)
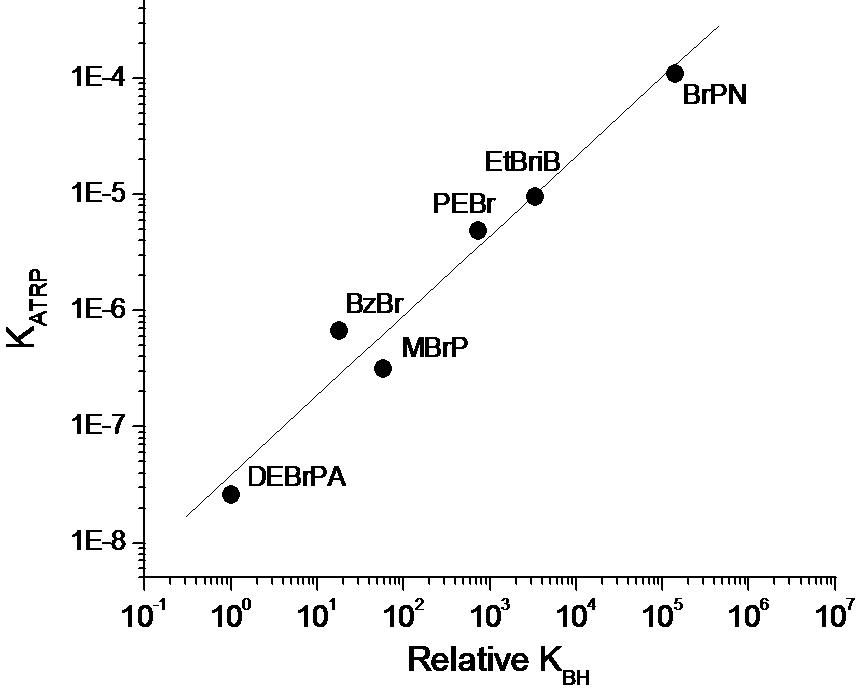
It was proposed that such calculations could also be used to predict equilibrium constants for dormant species derived from less reactive monomers. Knowing KBD as well as the rate constants of propagation for a given monomer, the rates of polymerization could be calculated for different monomers in ATRP under comparable conditions (same catalyst, constant KET, KEA, and KX). For example, if the ATRP of acrylonitrile reached 90% conversion in 1 second, methyl acrylate would require 2 hours, styrene 22 hours and vinyl acetate 30 years under the same conditions.(73) This calculation merely serves to demonstrate the necessity of choosing an appropriate catalyst and appropriate reaction conditions for each monomer.
Determination of the equilibrium and rate constants is crucial in order to understand the kinetics of an ATRP. Assuming steady-state kinetics, the rate of polymerization is given by:
![]()
This equation means that the rate of polymerization is controlled by the ratio of CuI to CuII and not the absolute amount of catalyst present in the reaction medium.
Understanding the implications of this equation was critical to the development of ARGET ATRP.(75) Experimentally, the values of KATRP can be determined by direct analysis of the polymerization mixture (by EPR,NMR, GC, GPC, IR...) or by the study of low molecular weight model compounds. Furthermore, while some side reactions (thermal-initiation of monomer, elimination reactions, transfer reactions, degradation of the catalyst...) and some physical parameters (viscosity, inhomogeneity...) may have an important effect on the kinetics of CRP the influence of these parameters may also be investigated by model studies or by computer simulation.(8,76-79)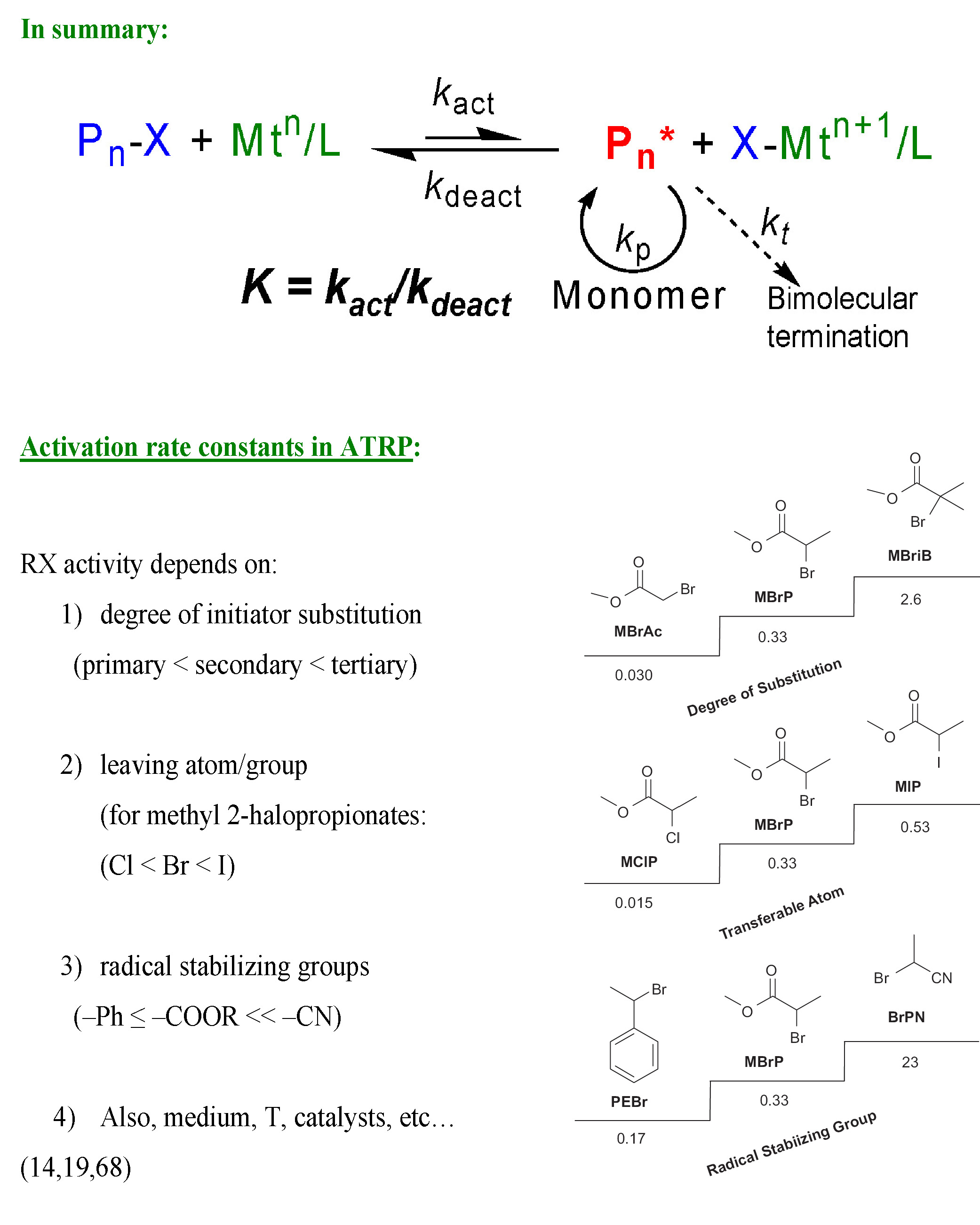
REFERENCES
(1) Minisci, F. Accounts of Chemical Research 1975, 8, 165-171.(2) Wang, J.-S.; Matyjaszewski, K. J. Am. Chem. Soc. 1995, 117, 5614-5615.
(3) Pintauer, T.; Matyjaszewski, K. Chem. Soc. Rev. 2008, 37, 1087-1097.
(4) Matyjaszewski, K. Macromol. Symp. 1998, 134, 105-118.
(5) Singleton, D. A.; Nowlan, D. T., III; Jahed, N.; Matyjaszewski, K. Macromolecules 2003, 36, 8609-8616.
(6) Fischer, H. Chemical Reviews 2001, 101, 3581-3610.
(7) Tang, W.; Tsarevsky, N. V.; Matyjaszewski, K. Journal of the American Chemical Society 2006, 128, 1598-1604.
(8) Vana, P.; Davis, T. P.; Barner-Kowollik, C. Macromolecular Rapid Communications 2002, 23, 952-956.
(9) Seeliger, F.; Matyjaszewski, K. Macromolecules 2009, 42, 6050-6055.
(10) Seeliger, F.; Matyjaszewski, K. Macromolecules 2010, 43, 5478.
(11) Morick, J.; Buback, M.; Matyjaszewski, K. Macromol. Chem. Phys. 2011, 212, 2423−2428.
(12) Mueller, L.; Jakubowski, W.; Matyjaszewski, K.; Pietrasik, J.; Kwiatkowski, P.; Chaladaj, W.; Jurczak, J. Eur. Polym. J. 2011, 47, 730-734.
(13) Braunecker, W. A.; Tsarevsky, N. V.; Gennaro, A.; Matyjaszewski, K. Macromolecules 2009, 42, 6348-6360.
(14) Tang, W.; Kwak, Y.; Braunecker, W.; Tsarevsky, N. V.; Coote, M. L.; Matyjaszewski, K. J. Am. Chem. Soc. 2008, 130, 10702-10713.
(15) Matyjaszewski, K.; Nanda, A. K.; Tang, W. Macromolecules 2005, 38, 2015-2018.
(16) Matyjaszewski, K.; Coca, S.; Gaynor, S. G.; Greszta, D.; Patten, T. E.; Wang, J.-S.; Xia, J. In USPTO; CMU: US5807937, 1998.
(17) Tsarevsky, N. V.; Pintauer, T.; Matyjaszewski, K. Macromolecules 2004, 37, 9768-9778.
(18) Matyjaszewski, K.; Coca, S.; Gaynor, S. G.; Wei, M.; Woodworth, B. E. Macromolecules 1998, 31, 5967-5969.
(19) Braunecker, W. A.; Matyjaszewski, K. Progress in Polymer Science 2007, 32, 93-146.
(20) Tsarevsky, N. V.; Braunecker, W. A.; Matyjaszewski, K. Journal of Organometallic Chemistry 2007, 692, 3212-3222.
(21) Matyjaszewski, K.; Xia, J. Chem. Rev. 2001, 101, 2921-2990.
(22) Kabachii, Y. A.; Kochev, S. Y.; Bronstein, L. M.; Blagodatskikh, I. B.; Valetsky, P. M. Polymer Bulletin 2003, 50, 271-278.
(23) Brandts, J. A. M.; van de Geijn, P.; van Faassen, E. E.; Boersma, J.; Van Koten, G. J. Organomet. Chem. 1999, 584, 246-253.
(24) Le Grognec, E.; Claverie, J.; Poli, R. J. Am. Chem. Soc. 2001, 123, 9513-9524.
(25) Maria, S.; Stoffelbach, F.; Mata, J.; Daran, J.-C.; Richard, P.; Poli, R. Journal of the American Chemical Society 2005, 127, 5946-5956.
(26) Uegaki, H.; Kotani, Y.; Kamigaito, M.; Sawamoto, M. ACS Symp. Ser. 2000, 760, 196-206.
(27) Kotani, Y.; Kamigaito, M.; Sawamoto, M. Macromolecules 2000, 33, 6746-6751.
(28) Kotani, Y.; Kamigaito, M.; Sawamoto, M. Macromolecules 1999, 32, 6877-6880.
(29) Matyjaszewski, K.; Wei, M.; Xia, J.; McDermott, N. E. Macromolecules 1997, 30, 8161-8164.
(30) Ando, T.; Kamigaito, M.; Sawamoto, M. Macromolecules 1997, 30, 4507-4510.
(31) O'Reilly, R. K.; Gibson, V. C.; White, A. J. P.; Williams, D. J. Polyhedron 2004, 23, 2921-2928.
(32) Teodorescu, M.; Gaynor, S. G.; Matyjaszewski, K. Macromolecules 2000, 33, 2335-2339.
(33) Ishio, M.; Katsube, M.; Ouchi, M.; Sawamoto, M.; Inoue, Y. Macromolecules 2009, 42, 188-193.
(34) Wang, Y.; Matyjaszewski, K. Macromolecules 2010, 43, 4003-4005.
(35) Kato, M.; Kamigaito, M.; Sawamoto, M.; Higashimura, T. Macromolecules 1995, 28, 1721-1723.
(36) Simal, F.; Demonceau, A.; Noels, A. F. Angew. Chem., Int. Ed. 1999, 38, 538-540.
(37) Kamigaito, M.; Watanabe, Y.; Ando, T.; Sawamoto, M. Journal of the American Chemical Society 2002, 124, 9994-9995.
(38) Haas, M.; Solari, E.; Nguyen, Q. T.; Gautier, S.; Scopelliti, R.; Severin, K. Advanced Synthesis & Catalysis 2006, 348, 439-442.
(39) Braunecker, W. A.; Itami, Y.; Matyjaszewski, K. Macromolecules 2005, 38, 9402-9404.
(40) Braunecker, W. A.; Brown, W. C.; Morelli, B. C.; Tang, W.; Poli, R.; Matyjaszewski, K. Macromolecules 2007, 40, 8576-8585.
(41) Percec, V.; Barboiu, B.; Neumann, A.; Ronda, J. C.; Zhao, M. Macromolecules 1996, 29, 3665-3668.
(42) Moineau, G.; Granel, C.; Dubois, P.; Jerome, R.; Teyssie, P. Macromolecules 1998, 31, 542-544.
(43) Wang, B.; Zhuang, Y.; Luo, X.; Xu, S.; Zhou, X. Macromolecules 2003, 36, 9684-9686.
(44) Granel, C.; Dubois, P.; Jerome, R.; Teyssie, P. Macromolecules 1996, 29, 8576-8582.
(45) Uegaki, H.; Kotani, Y.; Kamigaito, M.; Sawamoto, M. Macromolecules 1997, 30, 2249-2253.
(46) Uegaki, H.; Kamigaito, M.; Sawamoto, M. J. Polym. Sci., Part A: Polym. Chem. 1999, 37, 3003-3009.
(47) Moineau, G.; Minet, M.; Dubois, P.; Teyssie, P.; Senninger, T.; Jerome, R. Macromolecules 1999, 32, 27-35.
(48) Lecomte, P.; Drapier, I.; Dubois, P.; Teyssie, P.; Jerome, R. Macromolecules 1997, 30, 7631-7633.
(49) Tsarevsky Nicolay, V.; Matyjaszewski, K. Chem Rev 2007, 107, 2270-2299.
(50) Wang, Y.; Zhang, Y.; Parker, B.; Matyjaszewski, K. Macromolecules 2011, 44, 4022-4025.
(51) Mukumoto, K.; Wang, Y.; Matyjaszewski, K. ACS Macro Lett. 2012, 1, 599-602.
(52) Shinoda, H.; Matyjaszewski, K.; Okrasa, L.; Mierzwa, M.; Pakula, T. Macromolecules 2003, 36, 4772-4778.
(53) Gaynor, S. G.; Matyjaszewski, K. ACS Symp. Ser. 1998, 685, 396.
(54) Patten, T. E.; Xia, J.; Abernathy, T.; Matyjaszewski, K. Science 1996, 272, 866-868.
(55) Kwiatkowski, P.; Jurczak, J.; Pietrasik, J.; Jakubowski, W.; Mueller, L.; Matyjaszewski, K. Macromolecules 2008, 41, 1067-1069.
(56) Coca, S.; Jasieczek, C. B.; Beers, K. L.; Matyjaszewski, K. J. Polym. Sci., Part A: Polym. Chem. 1998, 36, 1417-1424.
(57) Qiu, J.; Charleux, B.; Matyjaszewski, K. Prog. Polym. Sci. 2001, 26, 2083-2134.
(58) Min, K.; Matyjaszewski, K. Central European Journal of Chemistry 2009, 7, 657-674.
(59) Matyjaszewski, K.; Gaynor, S. G.; Coca, S. In PCT Int. Appl.; (Carnegie Mellon University, USA). WO 9840415, 1998; p 230 pp.
(60) Matyjaszewski, K.; Beers, K. L.; Woodworth, B.; Metzner, Z. J. Chem. Educ. 2001, 78, 547-550.
(61) Jakubowski, W.; Matyjaszewski, K. Macromolecules 2005, 38, 4139-4146.
(62) Min, K.; Gao, H.; Matyjaszewski, K. Journal of the American Chemical Society 2005, 127, 3825-3830.
(63) Tang, H.; Shen, Y.; Li, B.-G.; Radosz, M. Macromol. Rapid Commun. 2008, 29, 1834-1838.
(64) Dong, H.; Matyjaszewski, K. Macromolecules 2008, 41, 6868-6870.
(65) Matyjaszewski, K. ACS Symp. Ser. 1998, 685, 258-283.
(66) Tsarevsky, N. V.; Tang, W.; Brooks, S. J.; Matyjaszewski, K. ACS Symposium Series 2006, 944, 56-70.
(67) Tsarevsky, N. V.; Braunecker, W. A.; Vacca, A.; Gans, P.; Matyjaszewski, K. Macromolecular Symposia 2007, 248, 60-70.
(68) Tang, W.; Matyjaszewski, K. Macromolecules 2007, 40, 1858-1863.
(69) Tang, W.; Matyjaszewski, K. Macromol. Theory Simul. 2008, 17, 359-375.
(70) Matyjaszewski, K.; Goebelt, B.; Paik, H.-j.; Horwitz, C. P. Macromolecules 2001, 34, 430-440.
(71) Pintauer, T.; Matyjaszewski, K. Coordination Chemistry Reviews 2005, 249, 1155-1184.
(72) Davis, K. A.; Matyjaszewski, K. Journal of Macromolecular Science, Pure and Applied Chemistry 2004, 41, 449-465.
(73) Gillies, M. B.; Matyjaszewski, K.; Norrby, P.-O.; Pintauer, T.; Poli, R.; Richard, P. Macromolecules 2003, 36, 8551-8559.
(74) Pintauer, T.; McKenzie, B.; Matyjaszewski, K. ACS Symposium Series 2003, 854, 130-147.
(75) Matyjaszewski, K.; Bombalski, L.; Jakubowski, W.; Min, K.; Spanswick, J.; Tsarevsky, N. In PCT Int. Appl.; (Carnegie Mellon University, USA). WO 2005087819, 2005; p 96 pp.
(76) Shipp, D. A.; Matyjaszewski, K. Macromolecules 1999, 32, 2948-2955.
(77) Shipp, D. A.; Matyjaszewski, K. Macromolecules 2000, 33, 1553-1559.
(78) Knuehl, B.; Pintauer, T.; Kajiwara, A.; Fischer, H.; Matyjaszewski, K. Macromolecules 2003, 36, 8291-8296.
(79) Pintauer, T.; Braunecker, W.; Collange, E.; Poli, R.; Matyjaszewski, K. Macromolecules 2004, 37, 2679-2682.