Development of ATRP
Understanding the Mechanistic Parameters of an Atom Transfer Radical Polymerization (ATRP):
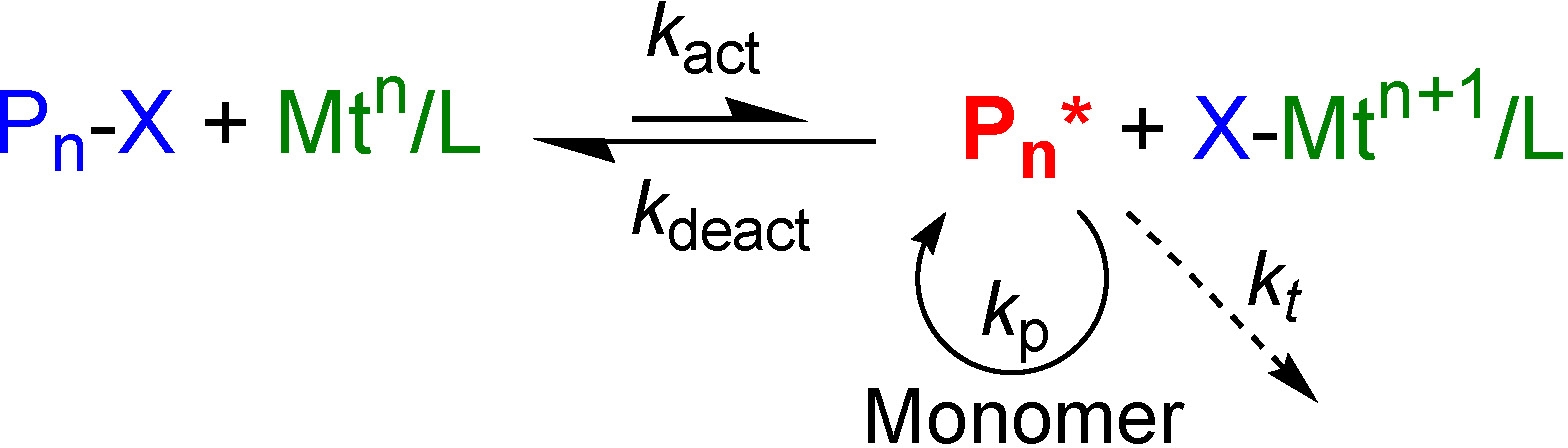
Details for the different procedures for initiation of an ATRP are provided on the initial page of how to conduct an ATRP. This section provides a short summary of the fundamental parameters that provide the foundation for this broadly applied controlled radical polymerization process.
The rate of an ATRP depends on the value of the equilibrium constant KATRP i.e. the ratio of rate constants for activation and deactivation. References cited below provide information on procedures used to determine these rate constants.
In 1995 it was established that the ATRP equilibrium, shown in the scheme above, can be approached from both sides:
- a standard or "normal" ATRP starting with the addition of the initiator R-X and transition metal complex Mtn/L (an ATRP initiator and a catalyst in a lower oxidation state) (1,2) to the reaction medium and,
- a "reverse" ATRP which starts by addition of the transition metal compound in its higher oxidation state, X-Mtn+1/L, which is then converted to the activator. In the initial example of a "reverse" ATRP (3,4) both the ATRP initiator and reduced "activator" catalyst complex were formed from radicals generated by decomposition of a standard free radical initiator.
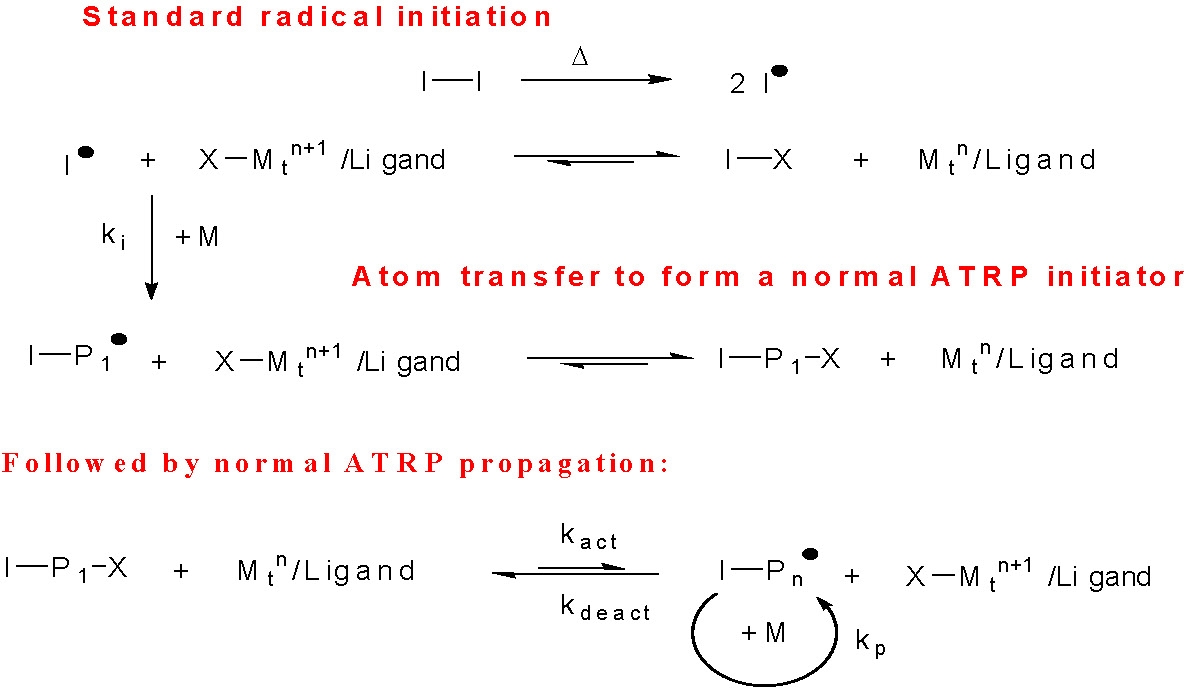
Successful polymerizations have been carried out starting with conventional free radical initiators, such as AIBN (5) and BPO (6) and higher oxidation state transition metal complexes.
- improvments to "reverse" ATRP, were subsequently developed to allow addition of an active transition metal complex in the oxidatively stable higher oxidation state, If an ATRP initiator is added to the reaction as a separate compound the higher oxidation state catalyst precursor for an active ATRP catalyst complex can be activated by adding a free radical initiator, Mt0, (7,8) or many other reducing agents. (9-13) The compounds directly added to the reaction are shown in red in the following scheme.
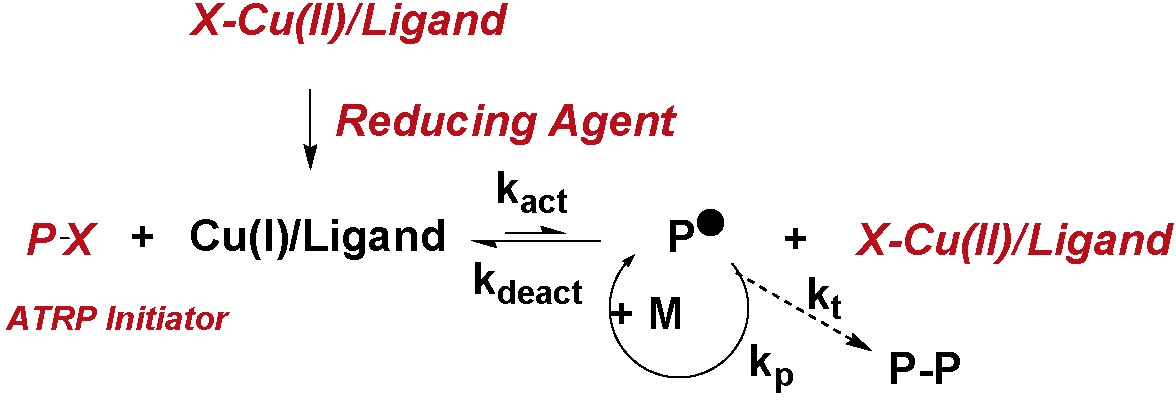
These non-radical forming reducing agents reduce the higher oxidation state transition metal complex, X-Mtm+1/Ln to form the X-Mtm/Ln activator in situ and allow formation of a pure copolymer. (14)
Note the formation, or addition, of Mt0 in any form to the reaction medium does not change the mechanism only the manner of attaining the equilibrium conditions and the rate of polymerization. (15,16)
There is abundant evidence that ATRP operates via a radical mechanism:
Including:
- Chemoselectivity,(17) cross propagation kinetics(18) and kinetic isotope effects(19) which are similar to the values seen for conventional radical polymerizations. Note however, the nature of a CRP with intermittent activation and deactivation can result in some deviation in cross propagation kinetics from those seen in a standard radical polymerization.(17,20).
- The polymerization is initially retarded by the presence of a small amount of oxygen as does the presence of true radical inhibitors, such as galvinoxyl and TEMPO that directly react with a growing radical and inhibit chain growth by terminating the reaction.(21) As shown below this provides a very convenient way to measure the activation rate constant for a specific ATRP catalyst system, initiator and solvent.
- On the other hand antioxidants, such as phenols, used to prevent in situ formation of peroxides which can initiate polymerization of (meth)acrylates, actually act to increase the rate of an ATRP reaction by reducing the concentration of the deactivator, X -Mtm+1.(10) Indeed, back in 1946, Walling (22) (who studied with Kharasch, (23) one of the earliest scientists to study radical addition reactions) warns against the misuse of the term "antioxidants", calling them "inhibitors", can lead to confusion about their mechanism of operation.
- The initial work on radical addition reactions, or Kharasch reactions,(24) was followed by the development of transition metal mediated atom transfer radical addition reactions.(25,26)
- ATRP is converted into a system which displays conventional radical polymerization characteristics upon the addition of octanethiol as a chain transfer reagent.(27) Chain transfer in butyl acrylate polymerization also resembles the conventional radical process.(28)
- ATRP can be carried out in the presence of water (29,30) or other protogenic reagents, and is tolerant of a variety of functionalities on the comonomers, just as in the case of standard radical polymerization reactions.(31)
- Moreover, the reactivity ratios, which are very sensitive to the nature of the active centers, are nearly identical to those reported for the conventional radical copolymerization of the same monomers but are different from those for anionic, group transfer, and cationic systems.(2,32-36)
- Regioselectivity and stereoselectivity are similar to, and do not exceed, that for a conventional radical polymerization. All the polymers formed by ATRP have regular head-to-tail structures with the dormant species of the typical secondary/tertiary alkyl halide structures, as evidenced by NMR. In addition, polymers have the similar tacticity as those prepared by a conventional free radical process.(20,21,36,37)
- A racemization study using optically active alkyl halides also supports the intermediacy of a radical.(21)
- EPR studies have revealed the presence of X-Mtn+1 species resulting from the persistent radical effect in normal ATRP reactions.(38-43)
- If a "classic" ATRP reaction is driven to high levels of monomer conversion a doubling of the molecular weight, or cross-linking in multi-functional initiator systems, has been observed as a result of radical-radical termination reactions.(44-46)
- Cross-exchange between different halogens (47) and different polymerization systems (thermal and ATRP or nitroxide mediated and ATRP) demonstrates they have the same intermediates and supports a common radical mechanism. An equimolar mixture of initiators for nitroxide-mediated polymerization and ATRP leads to the preparation of polystyrene with a unimodal molecular weight distribution (MWD).(48)
- Propagating free radicals have been observed directly by EPR in the polymerization of dimethacrylates.(49)
Activation rate constants (kact), for a specific ATRP reaction are typically determined from model studies in which the transition metal complex is reacted with a model alkyl halide in the presence of a radical trapping agent, such as TEMPO.(50) This works for every catalyst complex, even less active complexes based on pyridene imine ligands(51) and higher activity Me6TREN systems. The rates are typically determined by monitoring the rate of disappearance of the alkyl halide in the presence of excess activator (CuIX/Ln) and excess TEMPO, which traps radicals faster than CuIIX2/Ln deactivates them.(50)
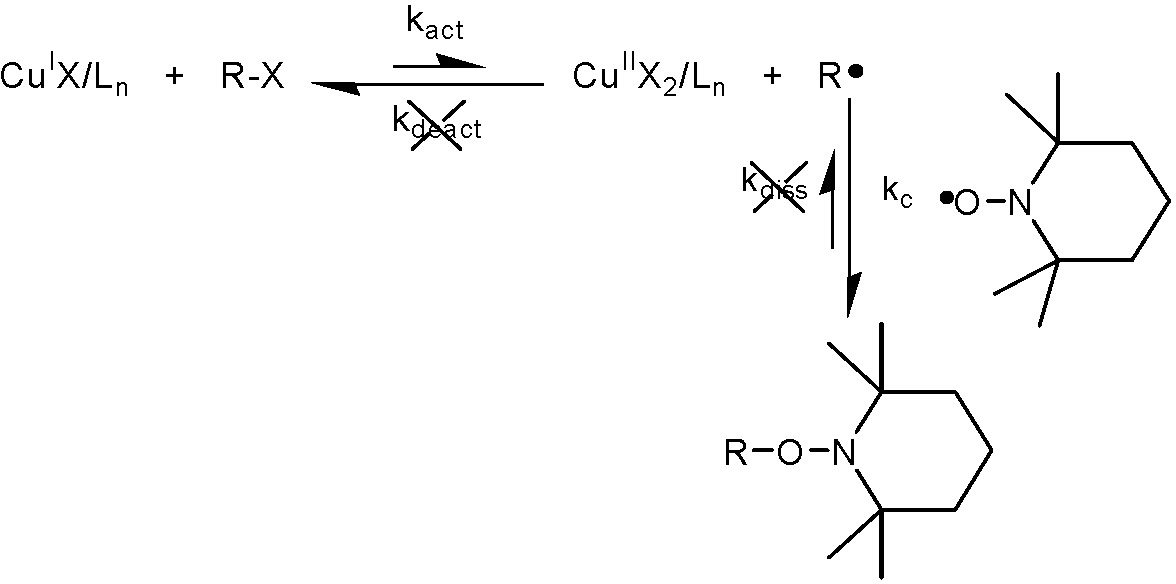
Under such conditions, the activation rate constant can be kinetically isolated from the deactivation rate constant and is given by ln([RX]0/[RX]t) = -kact[CuIX/Ln]0t .
An examination of the electrochemical properties of copper complexes was also conducted (50) to correlate the redox properties with the kinetic parameters of the activation and deactivation steps. The effect of ligand and initiator structure on the rate constants has also been examined. (56)
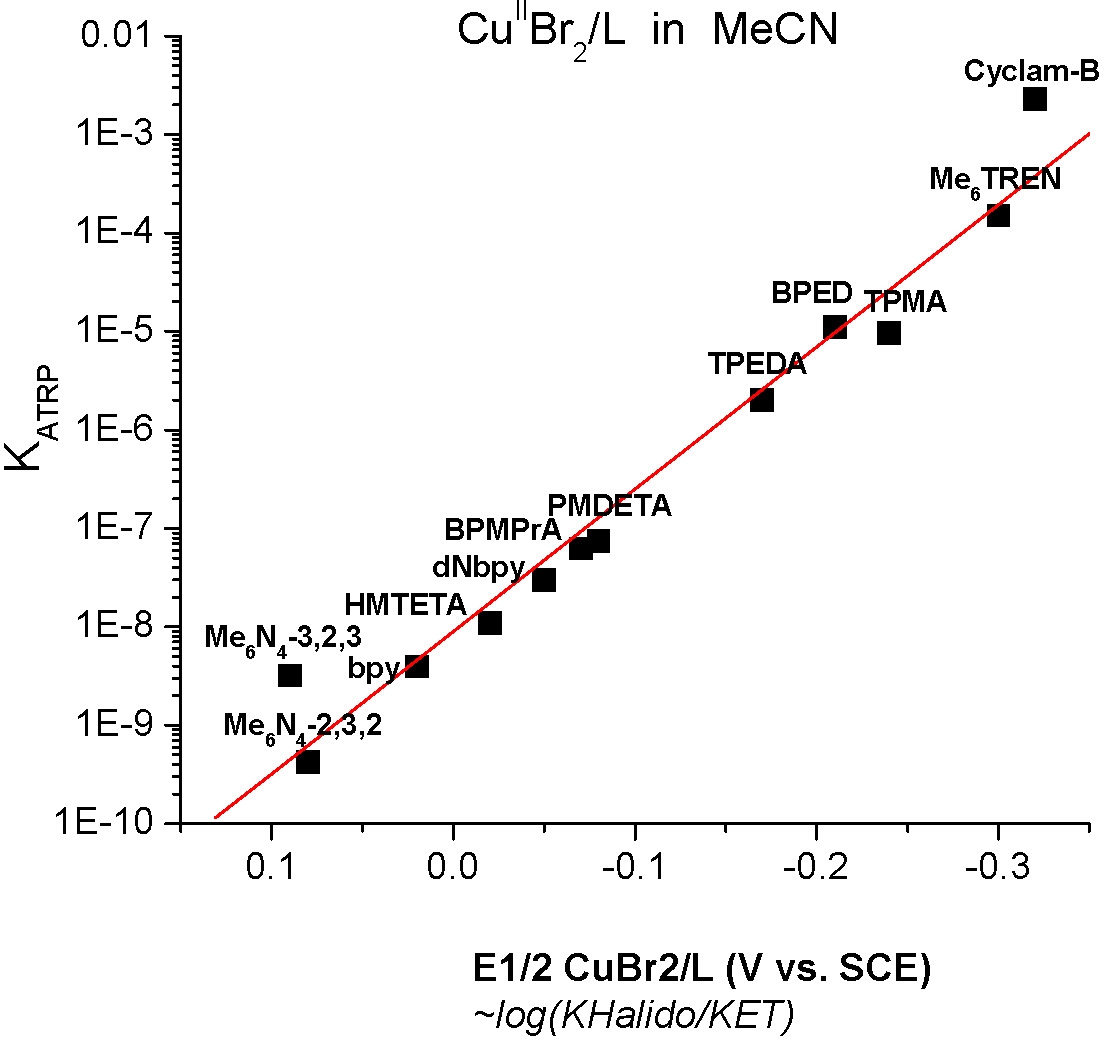
The values were also determined for some polymeric systems using GPC techniques and the results displayed similar kinetics.(52)
A novel approach towards screening of ATRP catalysts by electrospray ionization mass spectroscopy was reported. The ESI_MS methodology allows for the rapid assay of ATRP catalyst performance without prior polymerization experiments.(53,54) However, both methods are limited in measuring fast rate constants with the maximum upper limit of approximately ~2 M-1s-1. This limitation prompted a study of the stopped-flow technique for measuring very fast activation rate constants for model systems in copper-mediated ATRP,(55) since the mixing time (ca. 1 ms) and speed of data collection (one full spectrum ca. every 1 ms for diode-array UV-visible spectrophotometers) allow the measurement of pseudo-first order rate constants up to ca. 1.5.102 s-1 (t1/2 = ca. 5 ms) and second order rate constants up to ca. 1x104 M-1 s-1. The stopped-flow technique allowed determination of activation rate constants which were previously not accessible due to the limitations in measuring fast reaction rates with current GC and NMR techniques and demonstrated the effectiveness of the stopped-flow technique to measure fast reaction rates, opening up a new way to systematically determine activation rate constants and activation parameters for other highly active ATRP complexes. Results have expanded the range of both techniques and it is possible to push the upper limit of GPC techniques to ~10 M-1s-1 and stop flow measurements to 7.5x103 s-1.
The activity of the catalyst complexes formed with a broad spectrum of ligands have been determined and cover a wide range of activity; as detailed in the following pages and summarized in the interactive kinetic data section and in recent papers (52,56) and reviews on catalyst performance (57-59) that describe evaluation of catalyst complexes with different ligands displaying a range of activity exceeding 107.
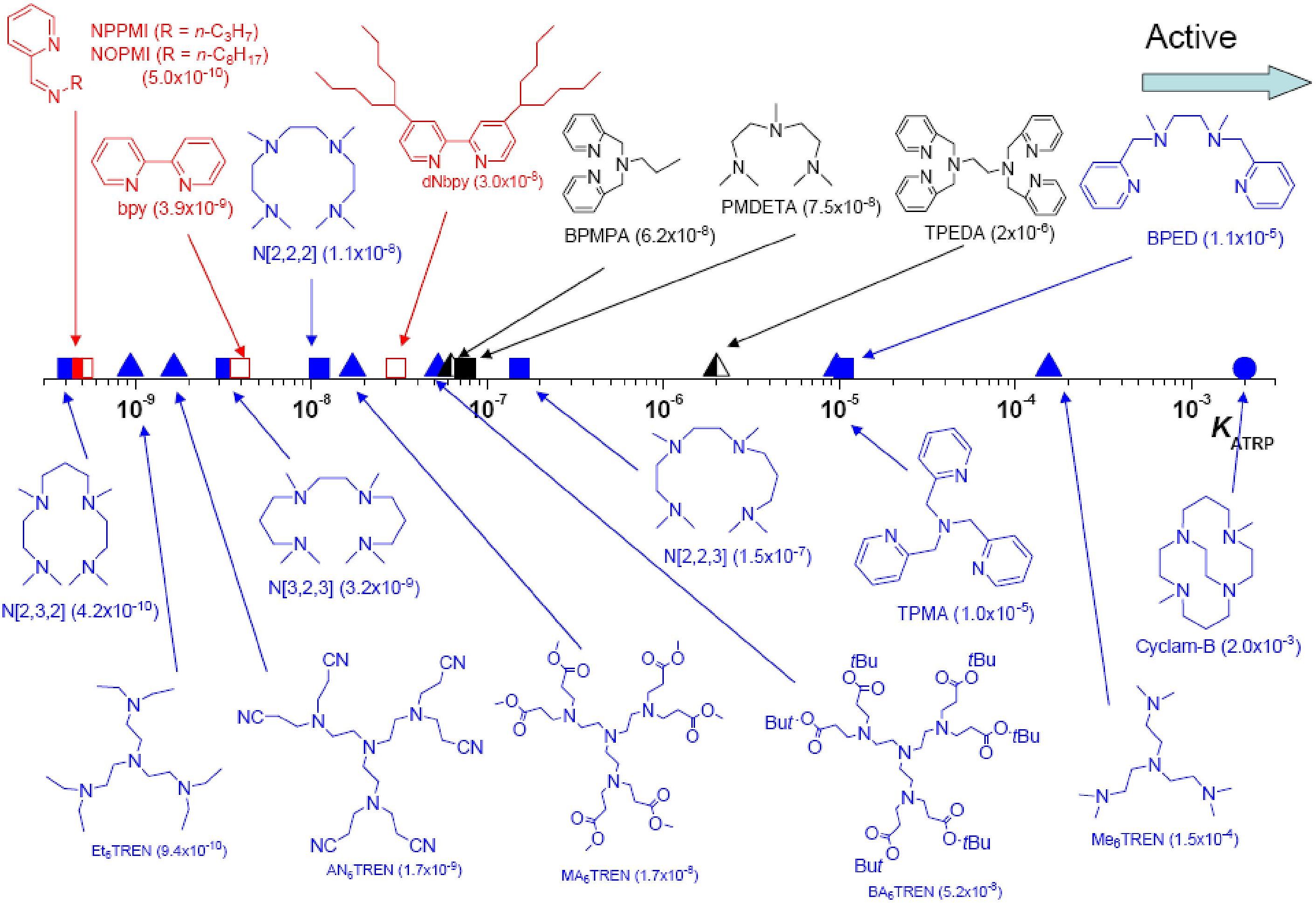
The above figure shows how keq for a series of copper based catalysts and how the activity changes with ligand.
The activation rate constants (kact) have been determined for a variety of initiators for Cu-mediated ATRP under the same conditions.(56,59,60)
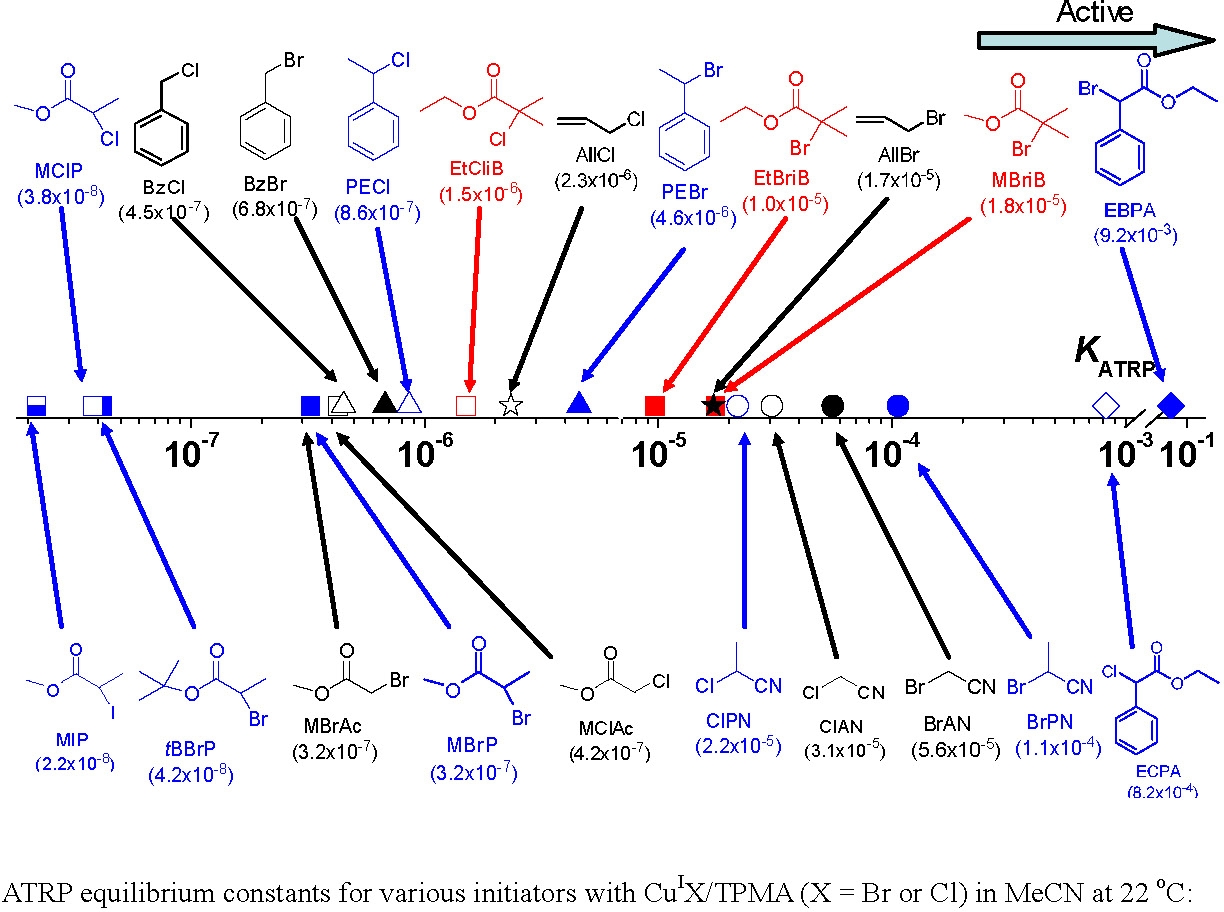
The ratio of the activation rate constants for the studied alkyl (pseudo)halides also exceeds 106 times.
REFERENCES
(1) Wang, J.-S.; Matyjaszewski, K. J. Am. Chem. Soc. 1995, 117, 5614-5615.
(2) Wang, J.-S.; Matyjaszewski, K. Macromolecules 1995, 28, 7901-7910.
(3) Wang, J.-S.; Matyjaszewski, K. Macromolecules 1995, 28, 7572-7573.
(4) Matyjaszewski, K.; Wang, J.-S. In PCT Int. Appl.; (Carnegie Mellon University, USA). WO 9630421, 1996; p 129 pp.
(5) Xia, J.; Matyjaszewski, K. Macromolecules 1997, 30, 7692-7696.
(6) Xia, J.; Matyjaszewski, K. Macromolecules 1999, 32, 5199-5202.
(7) Matyjaszewski, K.; Gaynor, S. G.; Coca, S. In PCT Int. Appl.; (Carnegie Mellon University, USA). WO 9840415, 1998; p 230 pp.
(8) Woodworth, B. E.; Metzner, Z.; Matyjaszewski, K. Macromolecules 1998, 31, 7999-8004.
(9) Gromada, J.; Matyjaszewski, K. Macromolecules 2001, 34, 7664-7671.
(10) Gnanou, Y.; Hizal, G. Journal of Polymer Science, Part A: Polymer Chemistry 2004, 42, 351-359.
(11) Jakubowski, W.; Matyjaszewski, K. Macromolecules 2005, 38, 4139-4146.
(12) Min, K.; Jakubowski, W.; Matyjaszewski, K. Macromolecular Rapid Communications 2006, 27, 594-598.
(13) Dong, H.; Matyjaszewski, K. Macromolecules 2008, 41, 6868-6870.
(14) Matyjaszewski, K.; Coca, S.; Gaynor, S. G.; Wei, M.; Woodworth, B. E. Macromolecules 1997, 30, 7348-7350.
(15) Tsarevsky, N. V.; Braunecker, W. A.; Matyjaszewski, K. Journal of Organometallic Chemistry 2007, 692, 3212-3222.
(16) Matyjaszewski, K.; Tsarevsky, N. V.; Braunecker, W. A.; Dong, H.; Huang, J.; Jakubowski, W.; Kwak, Y.; Nicolay, R.; Tang, W.; Yoon, J. A. Macromolecules 2007, 40, 7795-7806.
(17) Matyjaszewski, K. Macromolecules 1998, 31, 4710-4717.
(18) Haddleton, D. M.; Crossman, M. C.; Hunt, K. H.; Topping, C.; Waterson, C.; Suddaby, K. G. Macromolecules 1997, 30, 3992-3998.
(19) Singleton, D. A.; Nowlan, D. T., III; Jahed, N.; Matyjaszewski, K. Macromolecules 2003, 36, 8609-8616.
(20) Matyjaszewski, K. Macromolecules 2002, 35, 6773-6781.
(21) Matyjaszewski, K.; Paik, H.-j.; Shipp, D. A.; Isobe, Y.; Okamoto, Y. Macromolecules 2001, 34, 3127-3129.
(22) Walling, C.; Briggs, E. R. J. Am. Chem. Soc. 1946, 68, 1141-1145.
(23) Kharasch, M. S.; Jensen, E. V.; Urry, W. H. Science 1945, 102, 128.
(24) Dupont, G.; Dulou, R.; Pigerol, C. Compt. rend. 1955, 240, 628-629.
(25) Asscher, M.; Vofsi, D. Chem. Ind. (London) 1962, 209-210.
(26) Asscher, M.; Vofsi, D. Journal of the Chemical Society 1963, 1887-1896.
(27) Heuts, J. P. A.; Mallesch, R.; Davis, T. P. Macromol. Chem. Phys. 1999, 200, 1380-1385.
(28) Roos, S. G.; Muller, A. H. E. Macromol. Rapid Commun. 2000, 21, 864-867.
(29) Gaynor, S. G.; Qiu, J.; Matyjaszewski, K. Macromolecules 1998, 31, 5951-5954.
(30) Qiu, J.; Gaynor, S. G.; Matyjaszewski, K. Macromolecules 1999, 32, 2872-2875.
(31) Matyjaszewski, K.; Coca, S.; Nakagawa, Y.; Xia, J. Polym. Mater. Sci. Eng. 1997, 76, 147-148.
(32) Nishikawa, T.; Ando, T.; Kamigaito, M.; Sawamoto, M. Macromolecules 1997, 30, 2244-2248.
(33) Arehart, S. V.; Matyjaszewski, K. Macromolecules 1999, 32, 2221-2231.
(34) Matyjaszewski, K.; Ziegler, M. J.; Arehart, S. V.; Greszta, D.; Pakula, T. J. Phys. Org. Chem. 2000, 13, 775-786.
(35) Chambard, G.; Klumperman, B. ACS Symp. Ser. 2000, 768, 197-210.
(36) Ziegler, M. J.; Matyjaszewski, K. Macromolecules 2001, 34, 415-424.
(37) Takahashi, H.; Ando, T.; Kamigaito, M.; Sawamoto, M. Macromolecules 1999, 32, 6461-6465.
(38) Kajiwara, A.; Matyjaszewski, K.; Kamachi, M. Macromolecules 1998, 31, 5695-5701.
(39) Kajiwara, A.; Matyjaszewski, K. Macromol. Rapid Commun. 1998, 19, 319-321.
(40) Kajiwara, A.; Matyjaszewski, K. Macromolecules 1998, 31, 3489-3493.
(41) Matyjaszewski, K.; Kajiwara, A. Macromolecules 1998, 31, 548-550.
(42) Kajiwara, A.; Matyjaszewski, K. Polym. J. 1999, 31, 70-75.
(43) Kajiwara, A.; Matyjaszewski, K.; Kamachi, M. ACS Symp. Ser. 2000, 768, 68-81.
(44) Zhang, X.; Xia, J.; Matyjaszewski, K. Macromolecules 2000, 33, 2340-2345.
(45) Yoshikawa, C.; Goto, A.; Fukuda, T. e-Polymers 2002, Paper No. 13.
(46) Sarbu, T.; Lin, K.-Y.; Ell, J.; Siegwart, D. J.; Spanswick, J.; Matyjaszewski, K. Macromolecules 2004, 37, 3120-3127.
(47) Matyjaszewski, K.; Shipp, D. A.; Wang, J.-L.; Grimaud, T.; Patten, T. E. Macromolecules 1998, 31, 6836-6840.
(48) Korn, M. R.; Gagne, M. R. Chem. Commun. 2000, 1711-1712.
(49) Yu, Q.; Zeng, F.; Zhu, S. Macromolecules 2001, 34, 1612-1618.
(50) Matyjaszewski, K.; Paik, H.-j.; Zhou, P.; Diamanti, S. J. Macromolecules 2001, 34, 5125-5131.
(51) Tang, W.; Nanda, A. K.; Matyjaszewski, K. Macromolecular Chemistry and Physics 2005, 206, 1171-1177.
(52) Tang, W.; Matyjaszewski, K. Macromolecules 2006, 39, 4953-4959.
(53) di Lena, F.; Matyjaszewski, K. Chem. Commun. 2008, 6306-6308.
(54) di Lena, F.; Matyjaszewski, K. Dalton Transactions 2009, 8878-8884.
(55) Pintauer, T.; Braunecker, W.; Collange, E.; Poli, R.; Matyjaszewski, K. Macromolecules 2004, 37, 2679-2682.
(56) Tang, W.; Kwak, Y.; Braunecker, W.; Tsarevsky, N. V.; Coote, M. L.; Matyjaszewski, K. J. Am. Chem. Soc. 2008, 130, 10702-10713.
(57) Tsarevsky, N. V.; Matyjaszewski, K. Chemical Reviews 2007, 107, 2270-2299.
(58) Tsarevsky, N. V.; Braunecker, W. A.; Vacca, A.; Gans, P.; Matyjaszewski, K. Macromolecular Symposia 2007, 248, 60-70.
(59) Tang, W. In Chemistry of Synthetic High Polymers; Carnegie-Mellon Univ.: Pittsburgh,PA,USA, 2008; p 304 pp.
(60) Tang, W.; Matyjaszewski, K. Macromolecules 2007, 40, 1858-1863.