Chain extension
-
Halogen Exchange
-
"Equivalent" of "Halogen Exchange" in an ARGET Chain Extension:
Halogen Exchange
Halogen exchange is recommended for the preparation of block copolymers when one is moving from a macroinitiator of lower activity, such as a styrene or an acrylate, to continue the polymerization with a monomer that forms a more reactive dormant species, such as a methacrylate or acrylonitrile if one desires to obtain a narrow polydispersity for the second block.(1-9) The need for halogen exchange during an ATRP chain extension reaction arises as a consequence of a mismatch in reactivity when crossing over from terminal polymeric secondary alkyl halides to add monomers that form tertiary alky halides since propagation from the low fraction of initially formed tertiary terminal chain ends is faster that initiation from the remaining secondary alkyl halides. This miss-match in rate of initiation resulting in low cross-propagation initiation efficiency.
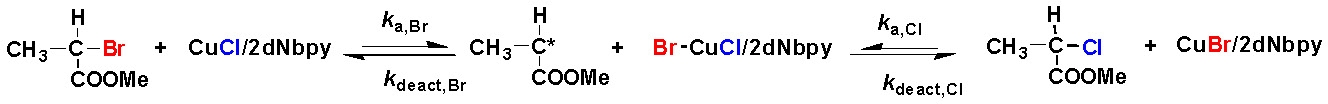
In a typical halogen exchange experiment, an An-Br macroinitiator is used but the catalyst complex is formed from CuCl rather than CuBr. In such a situation once the macro-radical An· is formed in the first activation step, it can add to the double bond of monomer B, yielding the radical An-B·, as in the previous case, but the newly formed radical can be deactivated by the CuII halide complex forming either a An-B-Br- or an An-B-Cl type dormant species. If the halogen exchange reaction is efficient the majority of the An-B· radicals are converted to An-B-Cl dormant species, while most of the An-type dormant species remain in the An-Br form.
The rational behind "halogen exchange" is that the value of the ATRP equilibrium constant for alkyl chloride-type of (macro)initiators is 1-2 orders of magnitude lower than the alkyl bromides with the same structure. These C-Cl bonds are activated more slowly, and thus the rate of propagation is decreased with respect to the rate of initiation from the added, more active macroinitiator, which effectively leads to increased initiation efficiency from the added macroinitiator and preparation of a second block with narrower polydispersity.
A well-defined A-b-B block copolymer will result in the case when KATRPB,Cl ´ kp < KATRPA,Br ´ ki.
The following GPC curves show chain extension of a polySty-Br macroinitiator with MMA using CuBr/dNbpy and CuCl/dNbpy as the catalyst. The reaction was conducted with the following ratio of reagents:
[MA]:[polySt-Br]:[CuX]:[dNbpy] = 500:1:1:2; at a temperature = 75oC.
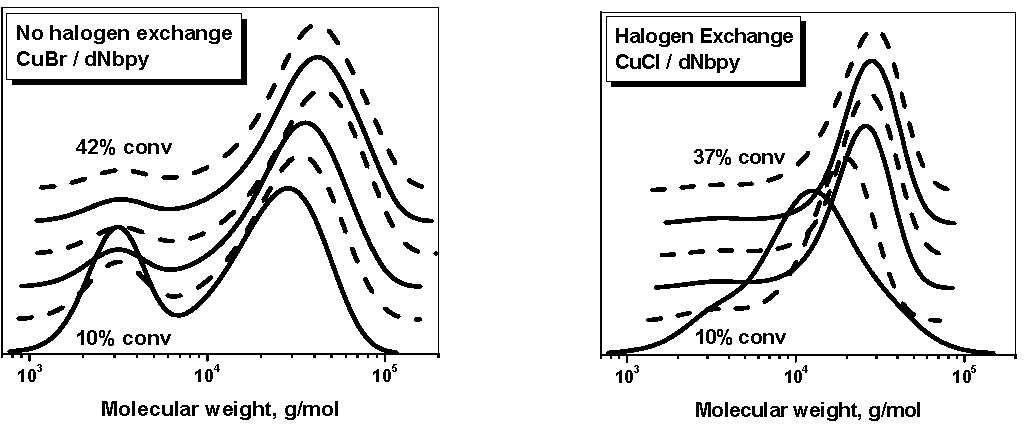
When the CuBr-based catalyst was used the faster activation of the newly formed polyMMA-Br combined with the more efficient addition of MMA-type radicals to MMA, compared to polySty-Br and Sty-type radicals, respectively, resulted in the preparation of copolymers with varying lengths of the polyMMA block mixed with unreacted macroinitiator. The use of CuCl/dNbpy as the catalyst significantly improved the cross-propagation kinetics and the MWD of the resulting copolymers was symmetrical. Moreover, the macroinitiator was practically completely consumed at relatively low MMA conversion.
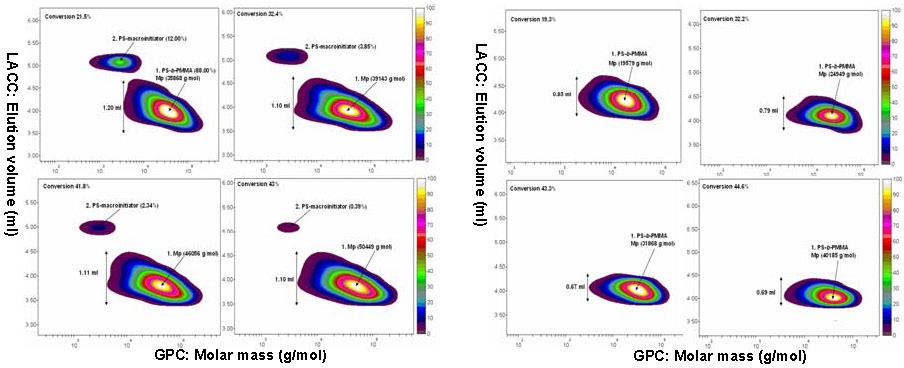
This is shown clearly in the following comparison of molecular heterogeneity between a block copolymer prepared with and without halogen exchange using Gradient Elution Chromatography (GPEC) and 2-Dimensional Chromatography. The effect of efficient cross-propagation is clearly shown by the clean shift in molecular weight and narrow molecular weight distribution. The 2D chromatographic analysis show that in the copolymer obtained using halogen exchange no macroinitiator remained at 19% MMA conversion, while ca. 10% unreacted polySty-Br macroinitiator was left in the reaction after 22% MMA conversion when halogen exchange was not employed.
The rate of halogen exchange depends strongly upon the nature of the ATRP catalyst and is higher when more active catalysts (catalysts high activation rate constant) are used. "Halogen Exchange" suffers from the same limitation present in standard "reverse" ATRP, namely that a stoiciometric amount of catalyst has to be added to convert each bromine chain end to a chain end containing a chlorine atom.
"Equivalent" of "Halogen Exchange" in an ARGET Chain Extension:
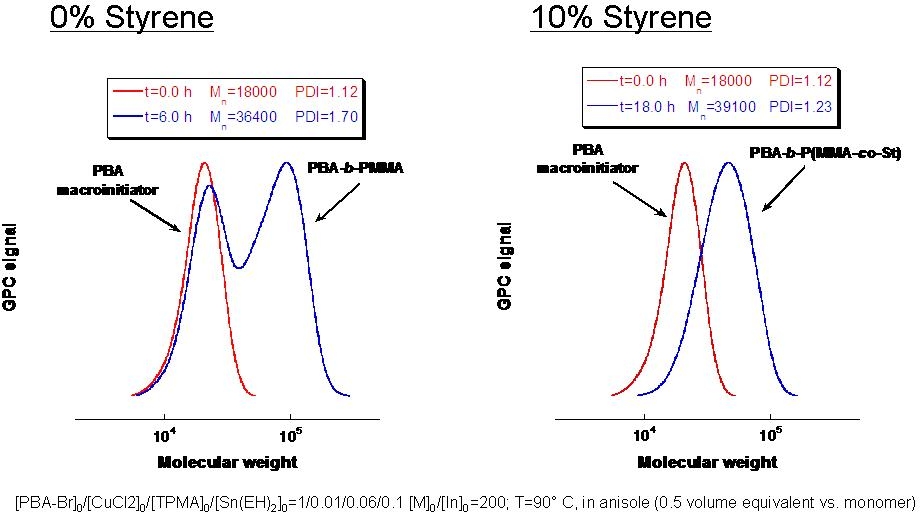
Since halogen exchange obviously can not be used for systems with a low concentration of catalyst successful chain extension of a polyacrylate or polystyrene macroinitiators with a methacrylate in an ARGET and ICAR ATRP presents a problem, The poor initiation efficiency of polystyrene and poly(butyl acrylate) macroinitiators during chain extension with methacrylates was resolved by employing a small amount of styrene as a comonomer in the second block.(10) Model studies with small molecule acrylate and styrene-based initiators determined that only 10 mol % of styrene was needed to provide sufficient control. Well-defined block copolymers poly(Bu acrylate)-b-poly(Me methacrylate-co-styrene) and polystyrene-b-poly(Me methacrylate-co-styrene) were synthesized using activators regenerated by electron transfer (ARGET) and initiators for continuous activator regeneration (ICAR) ATRP.
For example, starting from poly(Bu acrylate) macroinitiator (Mn = 18,000; Mn/Mw = 1.12), a poly(Bu acrylate)-b-poly(Me methacrylate-co-styrene) (Mn = 39,100; Mn/Mw = 1.23) was obtained. Without addition of styrene as a comonomer in the chain extension a block copolymer with a bimodal molecular weight distribution was formed: poly(Bu acrylate)-b-poly(Me methacrylate): Mn = 36 400, Mn/Mw = 1.70).
Computational simulations also indicated that the improved initiation efficiency was due to a ten fold higher concentration of CuI species in the copolymerization chain extension reaction caused by the lower KATRP of polystyrene chain ends.
REFERENCES
(1) Matyjaszewski, K.; Patten, T. E.; Xia, J. J. Am. Chem. Soc. 1997, 119, 674-680.
(2) Shipp, D. A.; Wang, J.-L.; Matyjaszewski, K. Macromolecules 1998, 31, 8005-8008.
(3) Matyjaszewski, K.; Shipp, D. A.; McMurtry, G. P.; Gaynor, S. G.; Pakula, T. J. Polym. Sci., Part A: Polym. Chem. 2000, 38, 2023-2031.
(4) Hong, S. C.; Pakula, T.; Matyjaszewski, K. Macromolecular Chemistry and Physics 2001, 202, 3392-3402.
(5) Matyjaszewski, K.; Gaynor, S. G.; Paik, H.-j.; Pintauer, T.; Pyun, J.; Qiu, J.; Teodorescu, M.; Xia, J.; Zhang, X. In PCT Int. Appl.; (Carnegie Mellon University, USA). WO 0056795, 2000; p 200 pp.
(6) Jousset, S.; Qiu, J.; Matyjaszewski, K.; Granel, C. Macromolecules 2001, 34, 6641-6648.
(7) Tsarevsky, N. V.; Sarbu, T.; Goebelt, B.; Matyjaszewski, K. Macromolecules 2002, 35, 6142-6148.
(8) Tang, C.; Kowalewski, T.; Matyjaszewski, K. Macromolecules 2003, 36, 1465-1473.
(9) Ramakrishnan, A.; Dhamodharan, R. Macromolecules 2003, 36, 1039-1046.
(10) Mueller, L.; Jakubowski, W.; Tang, W.; Matyjaszewski, K. Macromolecules 2007, 40, 6464-6472.