Biphasic Catalyst Systems
Prior to the development of ARGET and ICAR several other procedures had been examined for reduction and removal of catalyst complexes from an ATRP reaction. They are included herein for education purposes and to provide the information required for further development of these robust polymerization procedures, since operating the reaction with ppm levels of catalyst may still be undesirable for certain applications.
Solid Support:
Since waste or discarded catalyst increases the cost of the final product, one of the first approaches to reducing catalyst concentration in the final polymer was development of heterogeneous catalyst systems. Catalyst recovery and reuse are important when residual transition metal complex is not desired in the final product. Solid surface-immobilized catalysts offered a convenient method for catalyst recovery, after the reaction had been completed. There was a great deal of interest in immobilizing homogeneous transition metal polymerization catalysts on solid supports for the purpose of recovery and reuse. In a batch polymerization process, the catalyst from such a heterogeneous system can be removed by simple decantation, filtration, or centrifugation and provides a route for efficient catalyst separation with a possibility to reuse the catalyst after reactivation.
However initial comparison of these methods with traditional homogeneous bulk or solution based ATRP showed that surface-immobilized catalysts often provide products with higher molecular weights than predicted and broader molecular weight distributions, and sometimes requiring longer reaction times.(1-6) The level of control over the polymerization was improved by introduction of soluble spacer groups between the support and the ligand.(4) Nevertheless these approaches offered limited control over the polymerization due to diffusion limitations, primarily the result of slow deactivation. The implication of this observation was immediately seen in the preparation of polymers exhibiting higher than target molecular weight and broader molecular weight distribution. Activation occurs readily when the initiator containing a radically transferable atom, most often a halide, diffuses to the immobilized catalyst on the surface of a separable particle, the halide is transferred from the polymer chain end to the immobilized catalyst and initiation of polymerization occurs. However, deactivation does not occur until the active chain diffuses back to the surface of the particle and encounters a tethered higher oxidation state transition metal complex. Diffusion to and from the solid surface results in poor control.
A Hybrid Catalyst System:
A "hybrid" catalyst system can overcome one aspect of this difficulty. Our initial hybrid catalyst was composed of an immobilized catalyst working in conjunction with a small amount of soluble catalyst, predominately present in the deactivator oxidation state.(7) Controlled polymerization of vinyl monomers over a ligand supported catalyst was achieved in the presence of a small amount of soluble catalyst, preferentially present in the higher oxidation state that accelerates the rate of deactivation of the growing radical. The major fraction of the activator state of transition metal complex is immobilized on solid carriers. When used in a batch polymerization, the catalyst was successfully removed by simple filtration after the polymerization, resulting in low concentration (ppm levels) of residual transition metal in the polymer.(3) The role of the soluble deactivator, which is selected to display a different (higher) redox potential than the supported catalyst complex, is to selectively deactivate the growing chain. After deactivating the growing radical the soluble CuI species diffuses to the supported catalyst complex, without a significant diffusion barrier, since it is a low molecular weight species and, due to the different redox potential, the soluble catalyst is rapidly reconverted to the deactivator, the CuII species, through a halogen exchange reaction with the immobilized catalyst. In this way a very low concentration of a soluble catalyst acts to shuttle the deactivator from the supported catalyst to the active growing polymer chain.
In a full scale commercial operation there would be a slow accumulation of CuII on the tethered catalyst and this would have to be reactivated in situ by measured addition of a reducing agent; either a standard free radical initiator, or a transition metal in the zero oxidation state or, with the current state of knowledge based on application of ARGET ATRP, another non-radical forming reducing agent. Selection of a reducing agent that does not completely reduce the soluble CuII species would allow continuous activation and continuous polymerization to occur simultaneously.
The feasibility of using supported catalysts in an ATRP to facilitate catalyst recovery and recycling was recently reviewed by Zhu.(8) He noted that historically these catalysts consist of catalytic sites that are covalently tethered to larger supporting particles and it was generally believed that non-hybrid supported ATRP systems was a surface-mediated polymerization process; i.e., both activation and deactivation reactions take place at the surface of the particles. However, recent experiments had shown that this may not be the case. A theoretical analysis testing the concept of surface-mediated ATRP that showed that deactivation at the surface is unlikely. The topological/geographic isolation of catalytic sites, rather than polymer diffusivity limitation, was primarily responsible for this infeasibility. A trace amount of free catalyst in solution that minimizes the physical isolation is essential for mediating supported ATRP. This analysis suggests that many successful reports of controlled polymerization using a supported catalyst actually depended on formation of a fortuitous “hybrid” catalyst system.
Other heterogeneous catalyst recovery systems include use of a ligand with a reactive substituent that allows adsorption of the most of the soluble catalyst complex onto a short silica column.(9) The catalyst can be adsorbed onto an ion exchange resin then recovered during regeneration. In order to demonstrate the full utility of this hybrid catalyst system we have prepared polymers with high chain end functionality and complex polymeric architectures such as block, graft or gradient copolymers using this hybrid catalyst concept.(10,11)
Other research groups have also addressed this problem and developed other viable approaches to reduce catalyst concentrations and/or improve control in an ATRP reaction. Brittain and co-workers examined JandaJel resins and showed that they provided increased solvent compatibility and site accessibility due to the flexible cross-linker. When they were used for the ATRP of MMA, and 2-(dimethylamino)ethyl methacrylate (DMAEMA) the polymerizations proceeded quickly to high conversion (>90%) and were well-controlled.(12) Brittain also examined the use of a polyethylene-ligand system which was soluble at the reaction temperature but separated out at lower temperature.(13)
An extension of this concept is the use of precipitons to induce the precipitation of the catalyst after the polymerization was complete.(14-16)
Shen and Zhu have examined silica supported catalysts in continuous polymerization systems(17-20) and more recently a reversible system using hydrogen mediated self assembly.(19) They also provided an excellent summary of extensive efforts to reduce the concentration of the transition metal in the final polymer and reported that fine silica particles can be used as an example of a concept that essentially provides a homogeneous catalysis system during the polymerization but is sufficiently heterogeneous for separation/recovery. The system has been demonstrated to provide a viable process for recyclable polymerization catalysts.(20)
Jones(21) has also examined CuBr/bpy catalyst complexes supported on very small particles for ATRP of MMA and found that the mobile covalently tethered catalyst complexes provided good control during the polymerization, with MW close to theoretical values and narrow PDI. The catalyst was reactivated by using AIBN as reducing agent. However, the work on AGET ATRP reported elsewhere on these pages indicates that other reducing agents, potentially lower cost agents, have now been identified for catalyst reactivation. Jones’ results indicated that not all of the CuII species was reduced to CuI and this was the reason for slower rates of polymerization during reuse; not catalyst leaching. Leaching experiments indicate that the tethered systems result in no detectable soluble copper species and that the majority of the catalytic transformations occur at sites tethered to the surface. In contrast, use of the physisorbed catalyst results in a substantial amount of leached copper species.
Although Zhu(22) described another approach to systems that provide homogeneous reaction systems for catalysis but heterogeneous systems for separation/recovery resulting in low absolute concentrations of the catalyst in the final product which was attained by physical desorption of a low concentration of catalyst (21 ppm) from the silica support. This work has been summarized in a paper describing the fundamentals and development of high-efficiency supported catalyst systems for atom transfer radical polymerization.(23)
Shen recently examined another approach to supported dispersible/recyclable catalyst through the use of magnetic nanoparticles which were used to support an ATRP catalyst for polymerization of MMA. The nanoparticle-supported catalyst mediated a living/controlled radical polymerization of MMA as effectively as unsupported catalysts particularly after the addition of 22 mol % of CuBr2. The polymer MW was well-controlled with an initiator efficiency of 0.85 and polydispersity lower than 1.2. The supported catalysts could be easily separated/isolated using an external magnetic field. The activity of the recycled catalyst was regenerated by copper metal or in-situ regeneration using reducing agents such as alkylamine or tinII compounds. Chain extension confirmed the livingness of the system and it was concluded that nanosized supports had significantly reduced the adverse effects of tethered catalysts.(24)
Now that some well-behaved immobilized ATRP catalyst systems have been reported, a shift in focus towards improving and optimizing current systems, and developing protocols for catalyst recycling should take precedence, hopefully leading to eventual widespread industrial practice.
A "soluble" hybrid catalyst system:
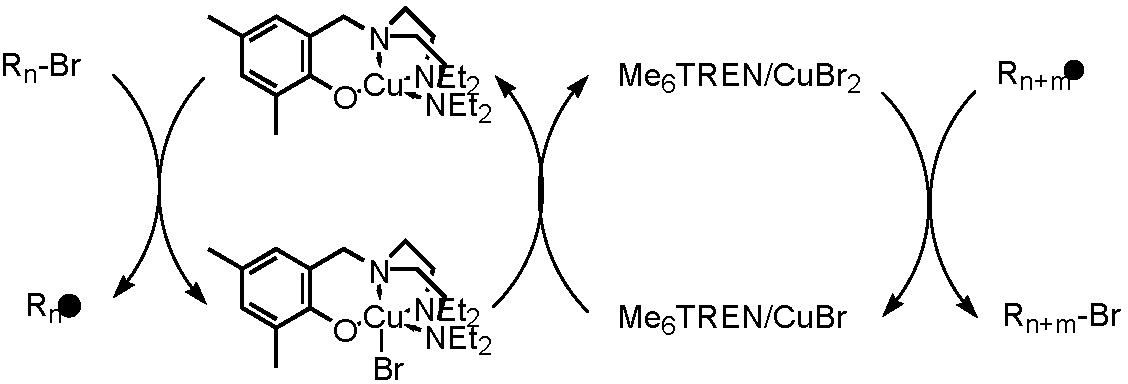
When examining the catalytic activity of a "halogen free" neutral copper complex, it was determined that CuI-phenoxide, the CuI species, activated the dormant chain but the CuII complex did not deactivate the growing chain. We took a page from the Hybrid Catalyst concept and added an efficient deactivator to the reaction. The CuI-phenoxide/(Me6TREN)CuIIBr2 system provides a highly active and controlled soluble hybrid catalytic system.(25)
REFERENCES
(1) Kickelbick, G.; Paik, H.-j.; Matyjaszewski, K. Macromolecules 1999, 32, 2941-2947.
(2) Haddleton, D. M.; Duncalf, D. J.; Kukulj, D.; Radigue, A. P. Macromolecules 1999, 32, 4769-4775.
(3) Hong, S. C.; Matyjaszewski, K. Macromolecules 2002, 35, 7592-7605.
(4) Shen, Y.; Zhu, S.; Zeng, F.; Pelton, R. H. Macromolecules 2000, 33, 5427-5431.
(5) Shen, Y.; Zhu, S.; Zeng, F.; Pelton, R. Macromol. Chem. Phys. 2000, 201, 1387-1394.
(6) Shen, Y.; Zhu, S.; Pelton, R. Macromolecules 2001, 34, 5812-5818.
(7) Hong, S. C.; Paik, H.-J.; Matyjaszewski, K. Macromolecules 2001, 34, 5099-5102.
(8) Faucher, S.; Zhu, S. Macromolecules 2006, 39, 4690-4695.
(9) Gromada, J.; Spanswick, J.; Matyjaszewski, K. Macromolecular Chemistry and Physics 2004, 205, 551-566.
(10) Hong, S. C.; Lutz, J.-F.; Inoue, Y.; Strissel, C.; Nuyken, O.; Matyjaszewski, K. Macromolecules 2003, 36, 1075-1082.
(11) Hong, S. C.; Neugebauer, D.; Inoue, Y.; Lutz, J.-F.; Matyjaszewski, K. Macromolecules 2003, 36, 27-35.
(12) Honigfort, M. E.; Brittain, W. J. Macromolecules 2003, 36, 3111-3114.
(13) Liou, S.; Rademacher, J. T.; Malaba, D.; Pallack, M. E.; Brittain, W. J. Macromolecules 2000, 33, 4295-4296.
(14) Honigfort, M. E.; Brittain, W. J.; Bosanac, T.; Wilcox, C. S. Macromolecules 2002, 35, 4849-4851.
(15) Honigfort, M. E.; Liou, S.; Rademacher, J.; Malaba, D.; Bosanac, T.; Wilcox, C. S.; Brittain, W. J. ACS Symposium Series 2003, 854, 250-266.
(16) Ayers, N.; Honigfort, M. E.; Brittain, W. J.; Wilcox, C. S. Polymer Preprints (Division of Polymer Chemistry) 2005, 46, 343-344.
(17) Shen, Y.; Zhu, S.; Pelton, R. Macromol. Rapid Commun. 2000, 21, 956-959.
(18) Shen, Y.; Zhu, S. AIChE Journal 2002, 48, 2609-2619.
(19) Yang, J.; Ding, S.; Radosz, M.; Shen, Y. Macromolecules 2004, 37, 1728-1734.
(20) Shen, Y.; Tang, H.; Ding, S. Progress in Polymer Science 2004, 29, 1053-1078.
(21) Nguyen, J. V.; Jones, C. W. Journal of Catalysis 2005, 232, 276-294.
(22) Faucher, S.; Zhu, S. Industrial & Engineering Chemistry Research 2005, 44, 677-685.
(23) Faucher, S.; Zhu, S. Journal of Polymer Science, Part A: Polymer Chemistry 2007, 45, 553-565.
(24) Ding, S.; Xing, Y.; Radosz, M.; Shen, Y. Macromolecules 2006, 39, 6399-6405.
(25) Inoue, Y.; Matyjaszewski, K. Macromolecules 2003, 36, 7432-7438.