Functional Biomaterials
Antibacterial Surfaces
Selective Protein Uptake
Polymer-Protein Conjugates
Polymer-DNA Biohybrids
Biodegradable Nanogels
Block Copolymers for Drug Delivery
Degradable (Nano)gels for Delivery Applications
Functional Star Molecules for Delivery Applications
Biolubrication (Wear protection without surface modicication)
Bioengineering Applications:
As noted throughout the web site one of the primary reasons for using a controlled radical copolymerization process for the preparation of a well defined macromolecule is that it is possible to incorporate site selected functional groups into the final copolymer. On this page we address how the group has worked with others to design materials that express specific bio-responsive properties, including antibacterial, protein conjugates, block copolymers designed for controlled drug delivery and materials designed for intra-cellular interactions.
Antibacterial Surfaces:
Grafting from:
Grafting onto:
Grafting from Magnetic Nanoparticles:
Grafting from:
Potentially harmful bacteria abound and pathogenic bacteria are frequently deposited onto surfaces that will be touched by many other people thereby spreading infection. It would be beneficial to have surfaces that kill bacteria on contact. Stable non-leaching antimicrobial surfaces are required since leaching systems, illustrated in the following schematic, are eventually exhausted, rendering the material ineffective and posing potential environmental risks to the surrounding environment.

Quaternary ammonium ion-containing polymers (PQA) are known to effectively kill cells and spores by disrupting cell membranes. Monomers, such as 2-dimethylaminoethyl methacrylate (DMAEMA), 4-vinyl pyridine (4-VP) and N-substituted acrylamides, that can be quaternized thereby providing biocidal activity, can be polymerized by ATRP. This means that stable non-leaching surfaces antimicrobial surfaces, shown in the following schematic, can be prepared by grafting from (1-3) or grafting onto surfaces. (4)
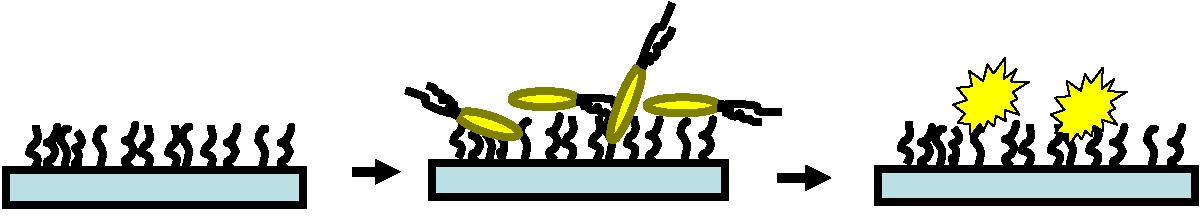
Covalent attachment of PQA via “grafting onto” or “grafting from” provides long-lasting biocidal activity with no danger to the environment. The effect of chain length, grafting density, composition, and architecture on biocidal activity is continuing to be examined and progress is summarized below.
Many surfaces have inherent functional groups that can be employed to conduct a “grafting from” reaction, the only requirement is the ability to tether initiators to the target substrate. In the case of paper and glass this is readily accomplished by reacting surface hydroxyl groups with 2-bromoisobutyryl bromide. (2) ATRP of DMAEMA followed by quaternization with ethyl bromide provided effective tethered biocidal functionality. When paper was treated the modified surfaces were extremely effective at killing E. coli, reducing the number of cells by four orders of magnitude, from 1.6 x 109 to 4.9 x 105, in one hour. The surface also showed activity against B. subtilis spores. The activity of a biocidal film on a glass surface survived repeated washing with aqueous detergent solution. Recent work by another group formed antibacterial silk (5) and cotton (6) by conducting DMAEMA grafting from ATRP reactions followed by quaternization. The modified fibers displayed good antibacterial properties and good resistance to removal of the reactive agent during laundry.
Grafting from:
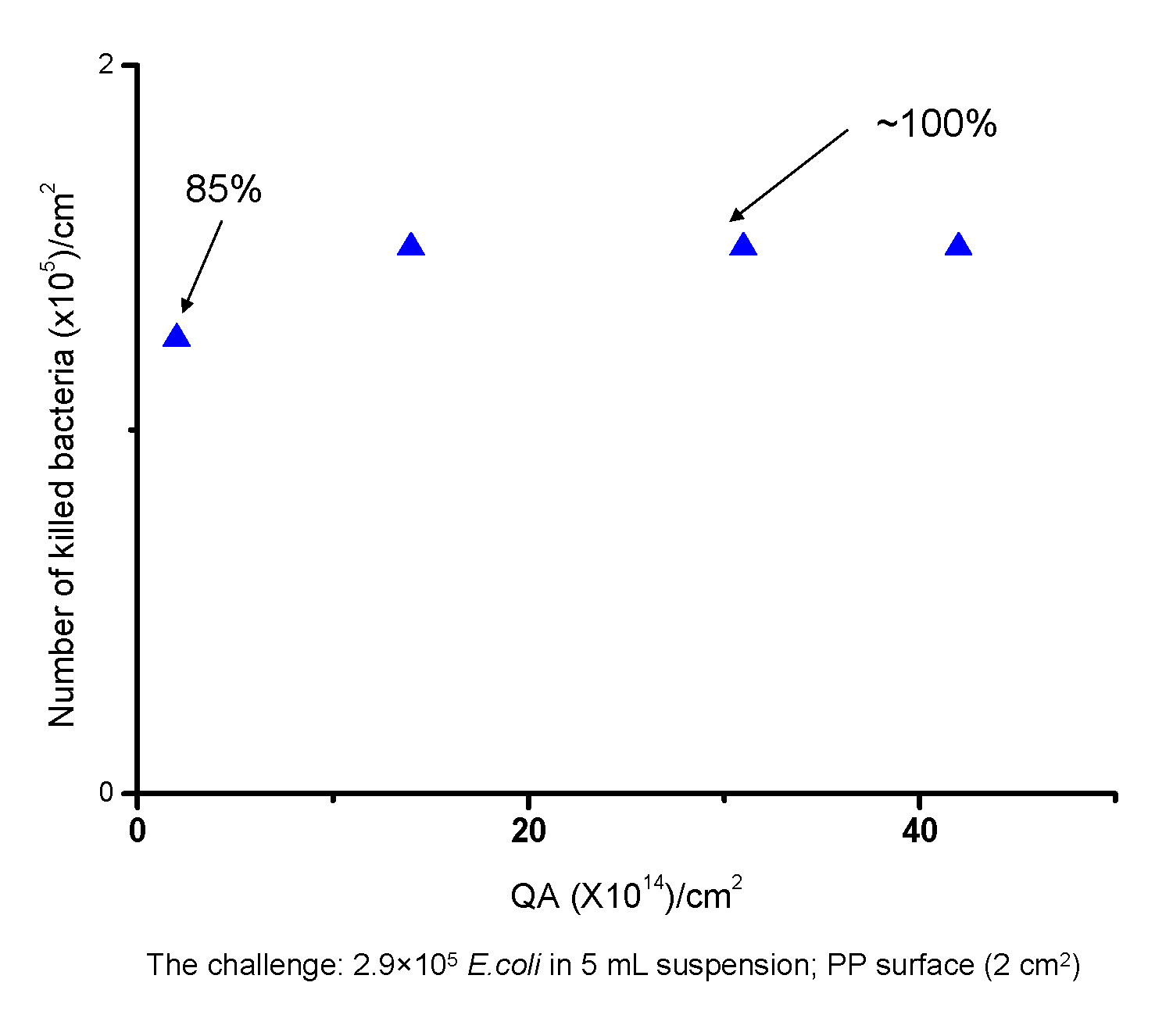
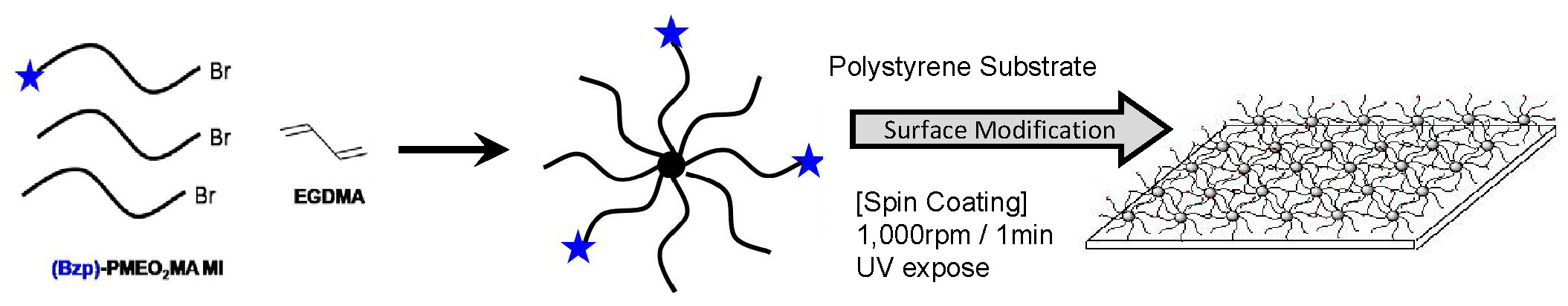
Polymeric surfaces are the most common surfaces encountered in everyday life and recent work has focused on preparing biocidal polymer surfaces. A non-leachable biocidal polypropylene (PP) surface was created by chemically attaching poly(quaternary ammonium) (PQA) chains to the surface of PP. As shown in the following schematic 2-benzophenonyl bromoisobutyrate was synthesized and spun-coated onto a polypropylene (PP) surface then exposed to UV light to form a benzophenone based radical that tethered the ATRP initiating functionality to the PP surface. A well-defined poly(2-(dimethylamino)ethyl methacrylate) (PDMAEMA), a precursor of a PQA, was grown from the surface of PP via ATRP in the presence of a sacrificial initiator which was used to estimate the MW of the attached chains. (3) The tertiary ammine groups in PDMAEMA were subsequently converted to a quaternary amine groups in the presence of ethyl bromide.
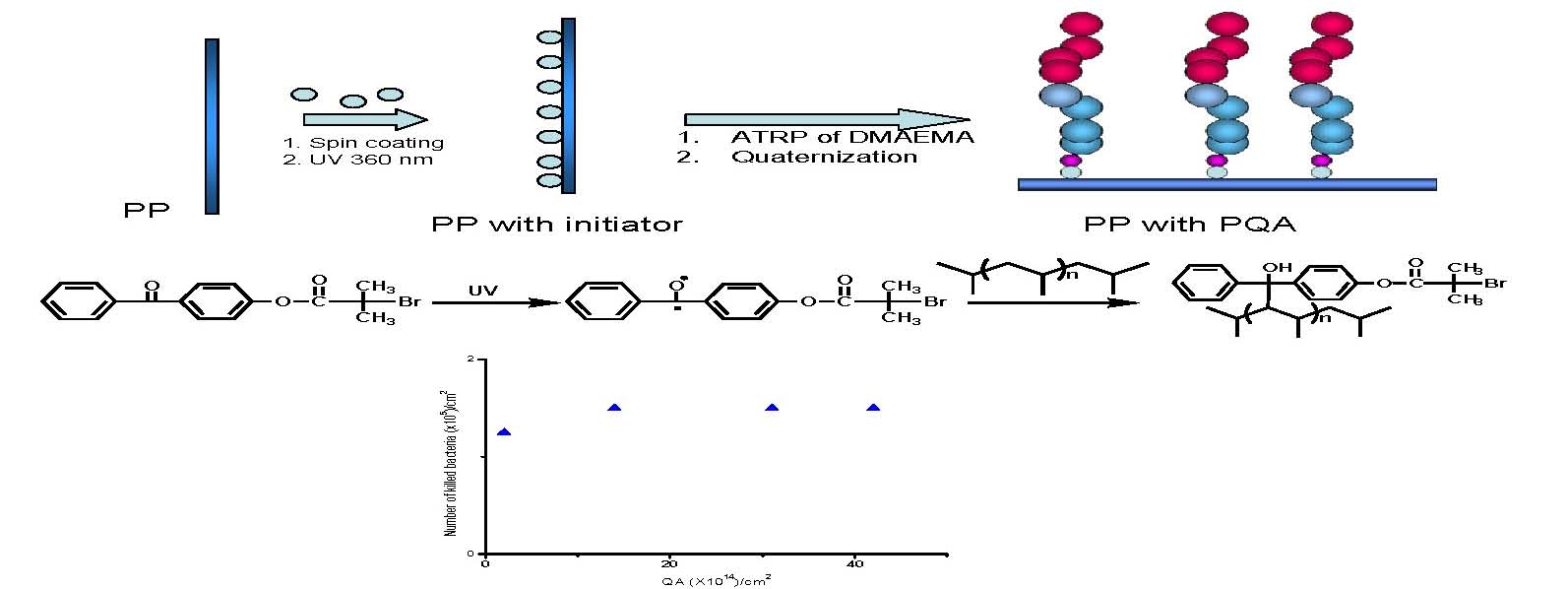
Successful surface modification was confirmed by ATR-IR, contact angle measurement, and an antibacterial activity test against Escherichia coli (E. coli), (see lower graph in schematic) showed that the biocidal activity of the resultant surfaces depends on the amount of the polymers grafted to the surface, i.e. the number of available quaternary ammonium units. Surfaces grafted with relatively high MW polymers (Mn > 10,000 g/mol) showed almost 100% killing efficiency, i.e. killing all of the added E. Coli (2.9 × 105) in the shaking test, whereas a lower biocidal activity (85%) was observed for the surface grafted with shorter PQA chains (Mn = 1,500 g/mol).
Successful surface modification was confirmed by ATR-FTIR, contact angle measurement, and an antibacterial activity test against Escherichia coli (E. coli). As shown in the lower figure in the above schematic the biocidal activity of the resultant surfaces depends on the amount of the polymers grafted to the surface (the number of available quaternary ammonium units). Surfaces grafted with relatively high MW polymers (Mn > 10,000 g/mol) showed almost 100% killing efficiency, i.e. killing all of the added E. Coli (2.9 x 105) in the shaking test, whereas a lower biocidal activity (85%) was observed for the surface grafted with shorter PQA chains (Mn = 1,500 g/mol).
It is envisioned that such a permanent, nonleaching biocidal surface treatment would find utility in food packaging facilities, household items and military applications.
Grafting onto:
While grafting from is an efficient method of tethering quaternizable polymers to a substrate a more convenient approach for existing household equipment, and even hospital use, would be a “consumer friendly” “grafting onto” approach such as spraying a solution of a reactive copolymer onto a surface. In order to demonstrate the ‘grafting onto’ approach well-defined block copolymers containing poly(2-dimethylaminoethyl methacrylate) PDMEAMA and poly(3-(trimethoxysilyl)propyl methacrylate (PTMSPMA) segments of different molecular weights and different backbone topology were prepared via ATRP and immobilized on a glass surface by simple immersion of the glass slide in an aqueous solution of the copolymer. (4) Immobilization of the block copolymer on the glass surface occurs via reaction between the PTMSPMA segment(s) and the surface silanol groups.
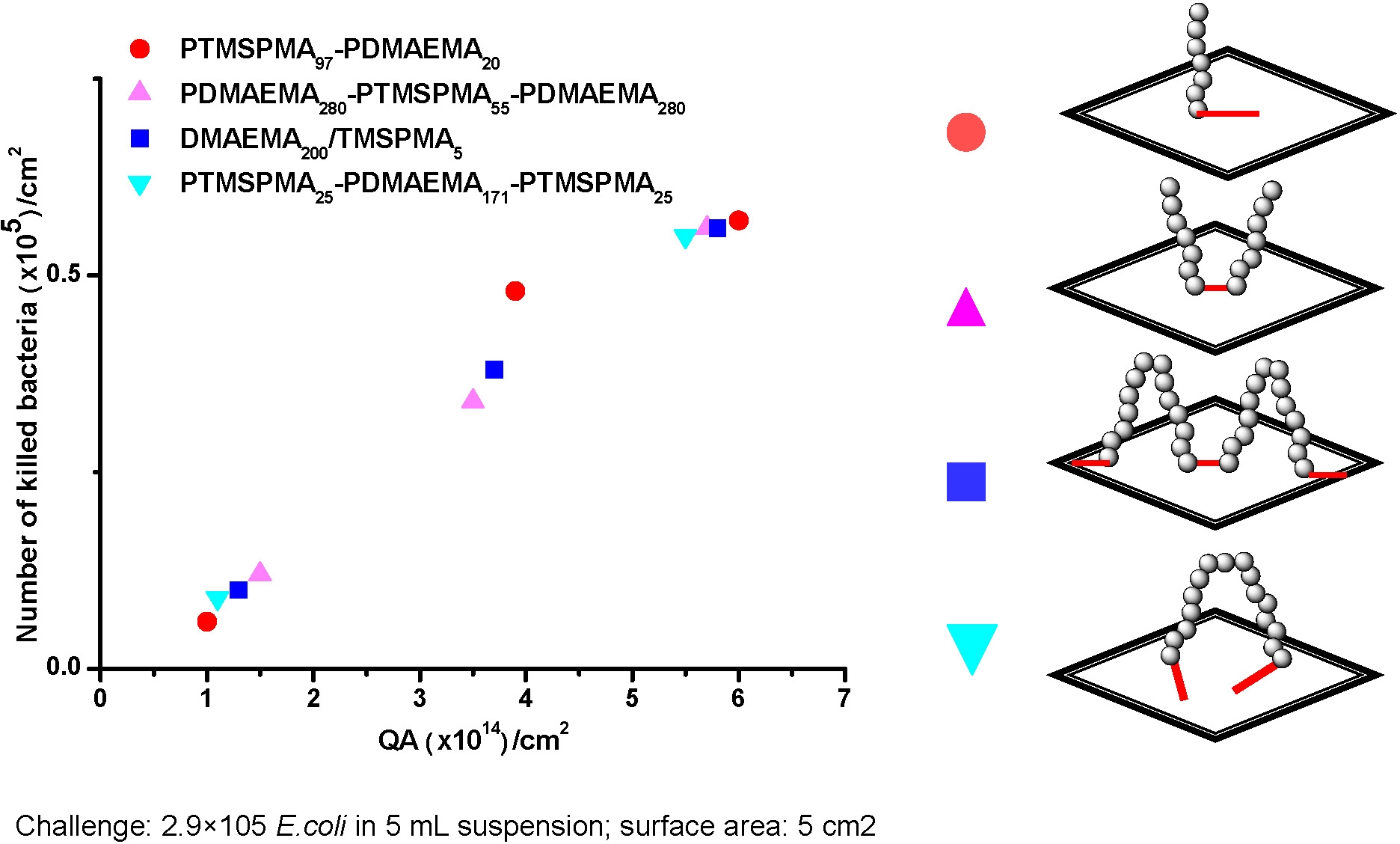
The efficiency of the tethering reaction was measured by addition of 1% of the sodium salt of fluorescein to the DMAEMA block prior to immersing the glass slide in the solution. Desorption of the dye after washing provided a measurement of the concentration of grafted chains on the surface. Grafting density was dependent on polymer concentration in the contacting solution and molecular weight of the copolymer as well as reaction time, with higher molecular weight copolymers providing lower graft density. However antibacterial activity depended only on the concentration of quaternary ammonium units tethered to the surface not on the polymer architecture.
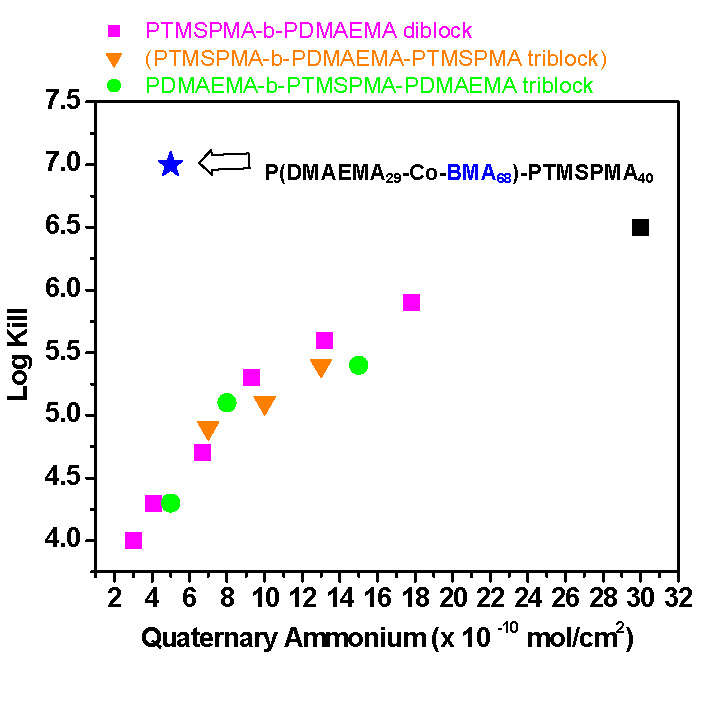
The interaction between bacteria and poly(quaternary ammonium) was investigated and the ability of the modified surfaces to kill bacteria was tested. Treated glass samples were incubated with a suspension of E. coli. The test results showed that the treated glass killed 104 ~ 106 of the contacting bacteria. Biocidal efficiency increased with the amount of quaternary ammonium units on the surface. Rather surprisingly introduction of hydrophobic units into a poly(quaternary ammonium) segment led to 100 times enhancement of biocidal activity with a log/kill of 7.0.
Modification of surfaces with tethered/tetherable polymers is applicable to a variety of surfaces. Control of surface morphology can be achieved by patterning the initiator layer and subsequent graft polymerization. Further variation of surface morphology can be achieved by phase separation on the nanoscale thereby providing precision surface modification.
A simple broadly applicable grafting to approach to form an antibacterial surface was developed by conducting an ATRP copolymerization of the precursor to an antibacterial monomer unit and a photoresponsive monomer, e.g. a monomer containing a benzophenone or xanthone functionality that can be modified after controlled radical polymerization to form a radical. A solution of the antibacterial/photoresponsive copolymer was deposited on a polymeric surface with an abstractable proton, such a polypropylene, and then exposure to light both crosslinks the deposited film and tethers it to the substrate.(7, 8)
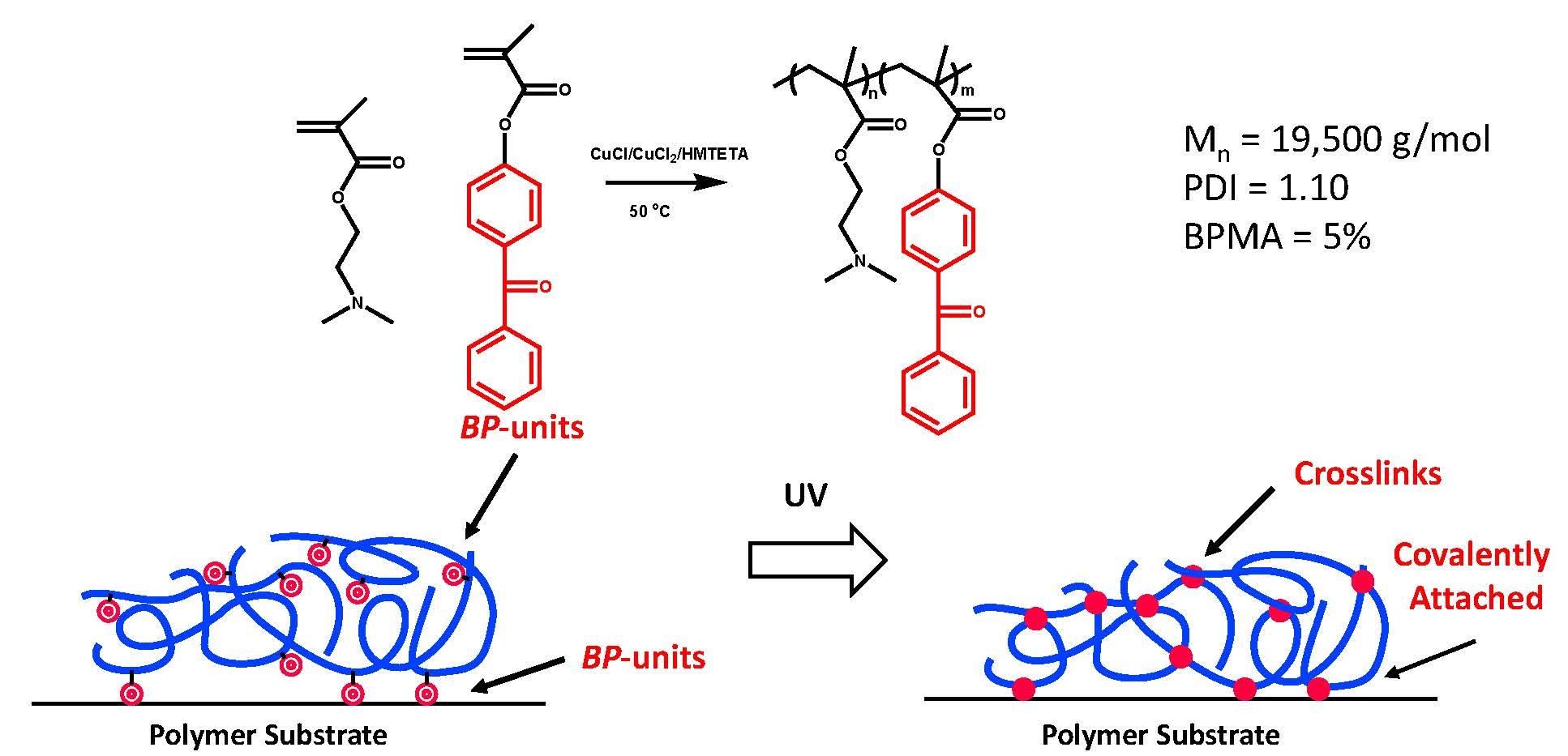
Since the copolymer is both water soluble and photoresponsive it can be deposited as an aqueous film on a surface and exposed to light, or if desired exposed to UV light through a photo-mask to form a patterned hydrogel.
Many different polymeric surfaces could be functionalized using similar chemistry and it is envisioned that such a permanent, nonleaching biocidal surface treatment would find utility in hospitals, food packaging facilities, household items and military applications.
Biocidal Magnetic Nanoparticles:
An extension of the use of preparing biocidal surfaces was the preparation of antibacterial magnetic particles via “grafting from”. (9) Highly efficient recyclable antibacterial magnetite nanoparticles consisting of a magnetic Fe3O4 core with an antibacterial poly(quaternary ammonium) (PQA) coating.
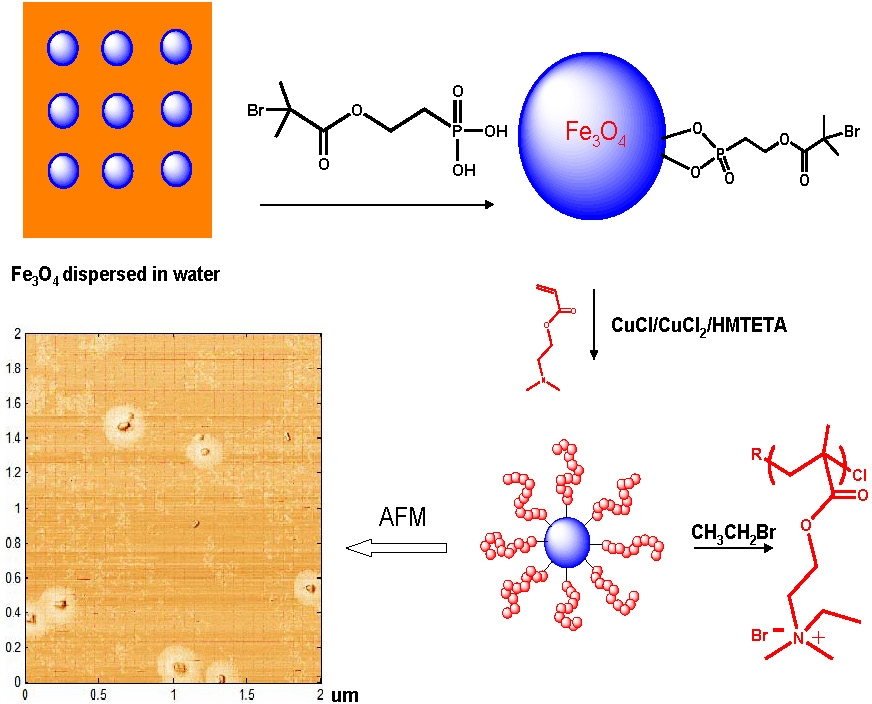
The concept was to use these particles to purify water, considering that the EPA indicated that 90% of the world’s water is contaminated. The advantages of using magnetic particles grafted with polymer chains that provide antibacterial properties over present procedures are:
a) that there would be no release of chemicals as the biocide is attached to the particle,
b) as indicated by the test determining killing efficiency the particles could kill bacteria with high efficiency due to the inherent large surface and,
c) after or during use the particles could be recycled by application of a magnetic field.
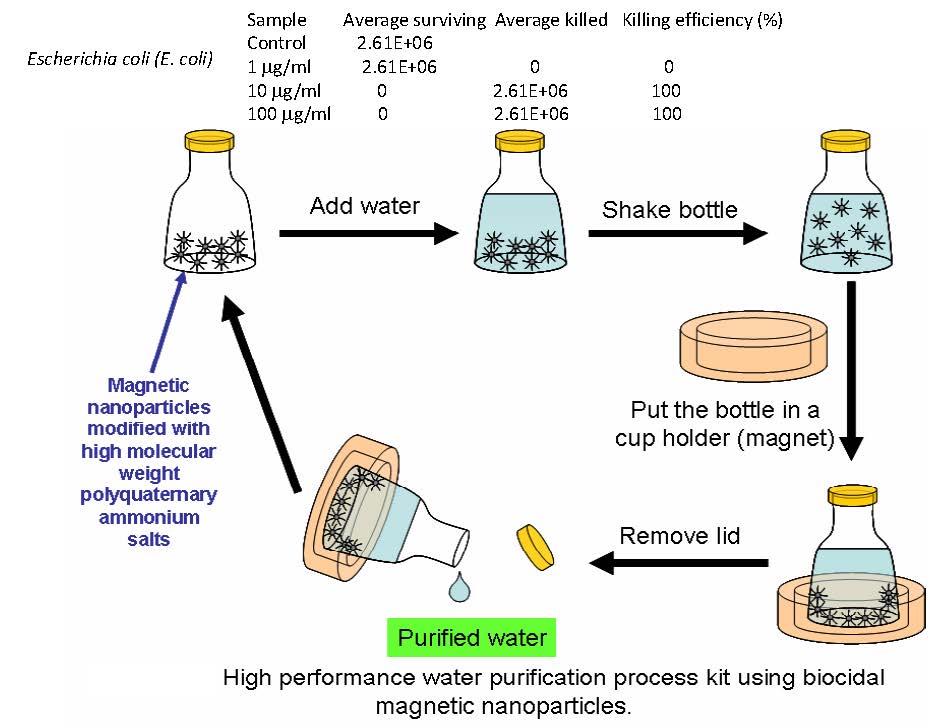
This was confirmed when the PQA-modified magnetite nanoparticles were dispersed in water and exhibited a response to an external magnetic field, making the nanoparticles easy to remove from water after antibacterial tests whose results are listed above the schematic. oparticles retained 100% biocidal efficiency against E. Coli (105 to 106 E. Coli/mg nanoparticles) during eight exposure/collect/recycle procedures without washing with any solvents or water.

The PQA-modified magnetite nanoparticles retained 100% biocidal efficiency against E. coli (105 to 106 E. coli/mg nanoparticles) during eight exposure/collect/recycle procedures without washing with any solvents or water. The exemplified small-scale procedure could be scaled-up to purify water in the trailor of a truck.
Another advantage of surface-initiated ATRP is that it allows one to precisely design the morphology and properties of the tethered polymer brushes. Indeed a gradient surface with varied grafting density or molecular weight has been successfully prepared by surface-initiated ATRP. (10)
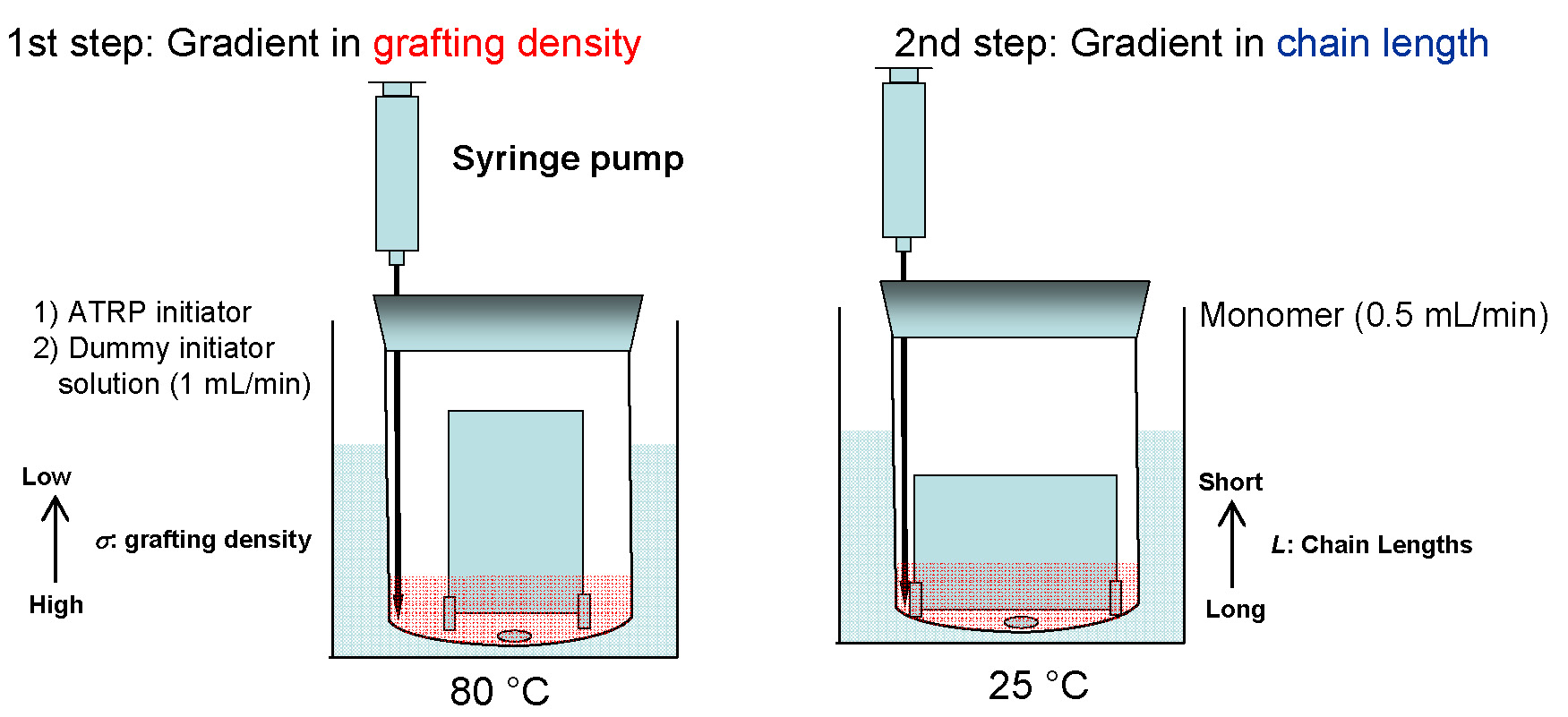
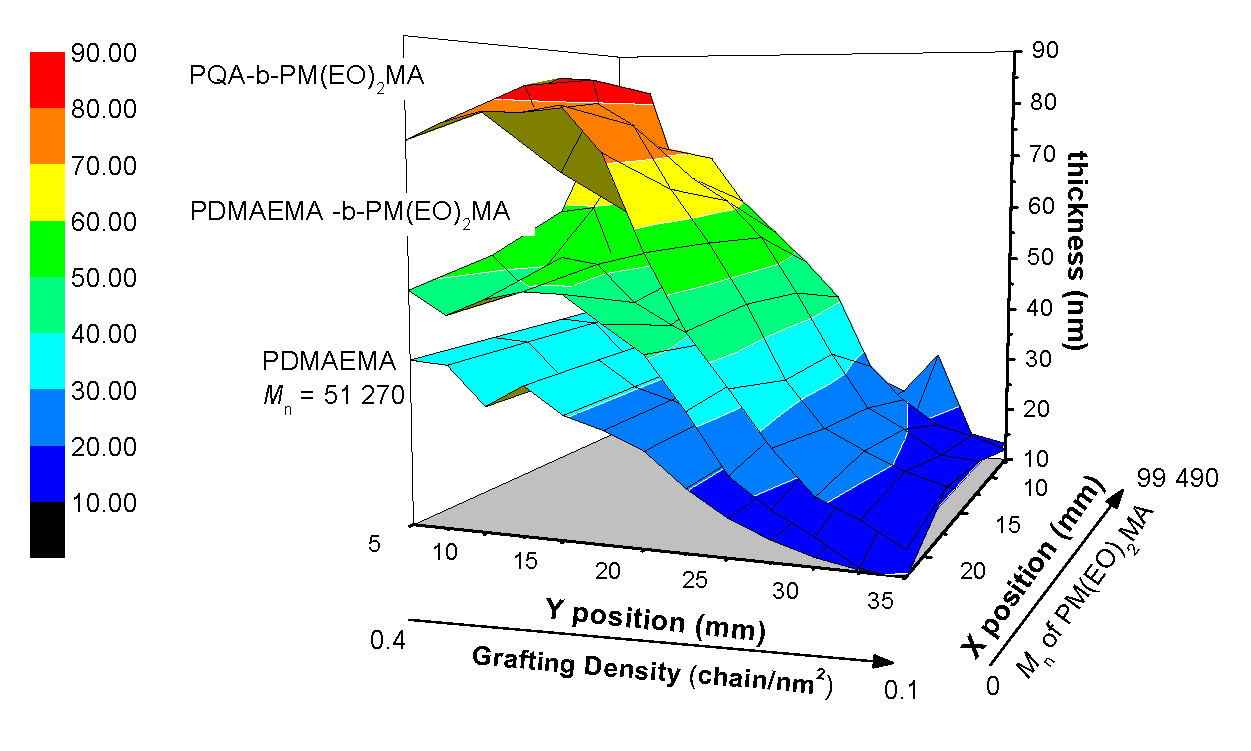
The dry layer thickness of the polymer brushes was controlled by polymerization time and/or initiator density on the surface.
This ability to control all aspects surface structure allows the properties of the antimicrobial polymer brushes to be rationally tailored. A combinatorial screening tool was developed to elucidate the role of chain length and chain density on cell kill in a single experiment.
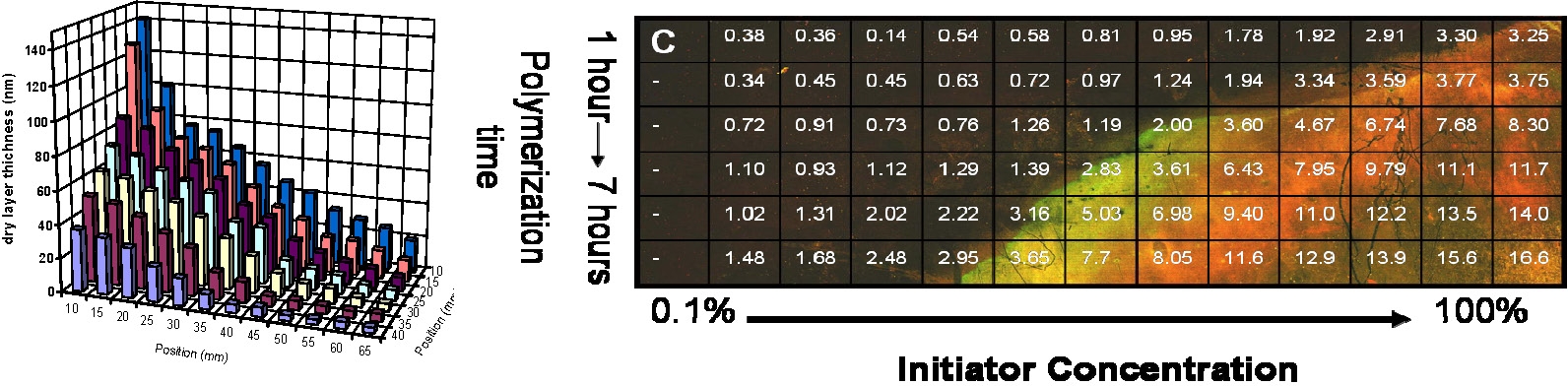
The results indicate that surface charge density, is a critical element in designing a surface for maximum kill efficiency. The biocidal surfaces that were most efficient had charge densities of greater than 1 - 5 X 1015 accessible quaternary amine units/cm2.
Biocidal surfaces are not the only type of bio-responsive surfaces that can be created from tethered poly(2-(dimethylamino)ethyl methacrylate) (PDMAEMA) brushes.
Selective Protein Uptake:
Surface plasmon resonance was used to measure binding of proteins from solution to poly(2-(dimethylamino)ethyl methacrylate) (PDMAEMA) brushes end-grafted from gold surfaces. (11) These brushes displayed a high capacity for electrostatically selective protein uptake. The net negatively charged protein BSA was taken up in amounts that approach its aqueous solubility limit in the case of PDMAEMA brushes at high grafting densities. These are among the highest reported protein binding capacities for ion exchange media. BSA binding scaled linearly with the mass of PDMAEMA grafted per unit area, with a constant ratio of approximately 120 DMAEMA monomer units per bound BSA molecule. The kinetics of BSA uptake in the brush is considerably more rapid than the slow asymptotic approach to adsorption saturation that is often seen for BSA adsorption to a solid surface. The high affinity for BSA was evident in the complete lack of desorption from the brush when rinsing with dilute NaCl solutions. BSA desorption from the brush required changes in solution pH and/or ionic strength to eliminate its net electrostatic attraction to PDMAEMA. Desorption could be achieved by using pH and/or ionic strength changes to interfere with the electrostatic attraction for BSA.
In contrast, PDMAEMA had no affinity for the net positively charged protein lysozyme. These results indicate that particles with tethered PDMAEMA chains can provide a charge selective, high capacity ion exchange medium for protein binding.
One important advantage of grafting from method via CRP, specifically ATRP, is the ease with which specific surfaces can be functionalized with initiating moieties for the preparation of well-defined high-density of polymer brushes. The highest grafting density can reach up to 1 chain/nm2. In that case, the polymer chain has a stretched conformation which is quite different from those of the semi-dilute polymer brushes previously studied, leading to very unique properties.
Polymer-Protein Conjugates:
The field of polymer bioconjugation (i.e. covalent attachment of synthetic polymers to biological entities such as nucleic acids, oligopeptides, proteins, enzymes, carbohydrates, viruses or cells) has evolved rapidly during the last decade. (12, 13) Polymer bioconjugates were initially developed by biochemists and had been exclusively studied for bio-medical applications. However, within the last few years the utility of this novel class of macromolecules has expanded and they are being examined by many groups in emerging areas of materials science. (14-22) Both the bio-component and the attached polymer based segment can span a range of complexity and functionality (23) and the synthesis of well-defined functional polymer bioconjugates has emerged as a central topic in macromolecular engineering.
A review paper which describes recent changes and progress in the field of bioconjugation; i.e. covalent attachment of synthetic polymers to biological entities such as nucleic acids, oligopeptides, proteins, enzymes, carbohydrates, viruses or cells, has been provided by two former group members. (16) The covalent conjugation approach can utilize either “grafting from” a tethered initiator (-I) or “grafting to” a functional group such as (-NH2) in which a preformed telechelic polymer is reacted with the targeted protein. (24)
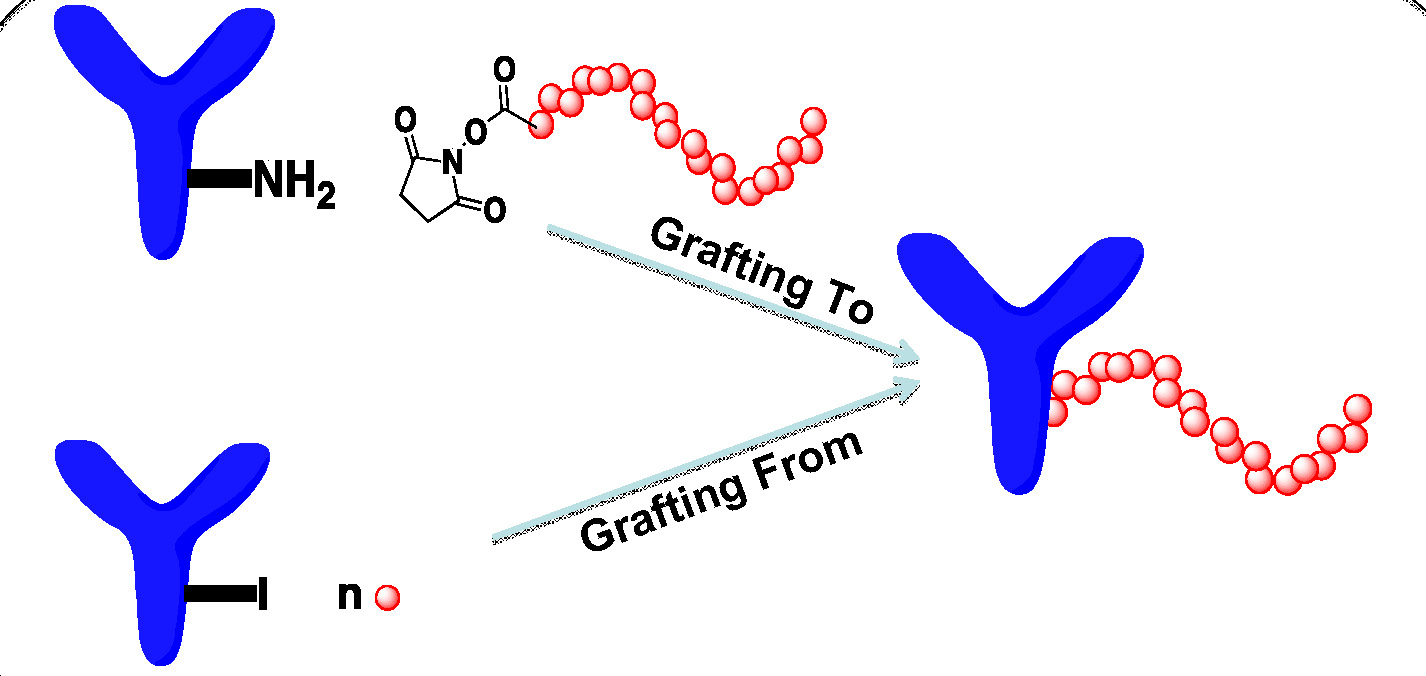
The advantage of prior “grafting to” techniques is the ability to form protein-polymer hybrid conjugates with predefined polymers while the disadvantages are that the formed bioconjugates are hard to purify and the grafting to reaction can generate non-uniform products. Nevertheless modern copper catalyzed synthetic methods such as ATRP and ‘‘click’’ chemistry coupling reactions have recently proven to be extremely versatile tools for the preparation of tailor-made polymer bioconjugates. (16, 25) One widely applied procedure for preparing such bioconjugates include preparation of functional telechelic polymers for direct attachment to the target bioresponsive molecules. (26) This procedure frequently employs transformation of terminal groups on polymers prepared by ATRP for subsequent click reactions. (27-29) In a more recent published collaboration with Román’s group in Spain smart heparin based bioconjugates were prepared using ATRP and click chemistry. (30) α-Azido functionalized PMDAETA and P(EO2MA-co-OEOMA300) polymers were prepared by AGET ATRP and the –COOH groups on bemiparin were reacted with propargylamine to generate alkynyl side groups. Click coupling was accomplished in the presence of CuSO4•5H2O and sodium ascorbate and the complex purified by dialysis. The bio-conjugates were both temperature and pH-responsive with the LCST values of the heparin bioconjugate scaling linearly with the composition of the copolymer segment.
The reverse approach where a targeted bio-responsive molecule is functionalized with an initiator for an ATRP, exemplified below by Streptavidin, followed by “grafting from” to tether a copolymer of desired composition and molecular weight to the functional bio-molecule also receiving increased levels of attention. (31-33)
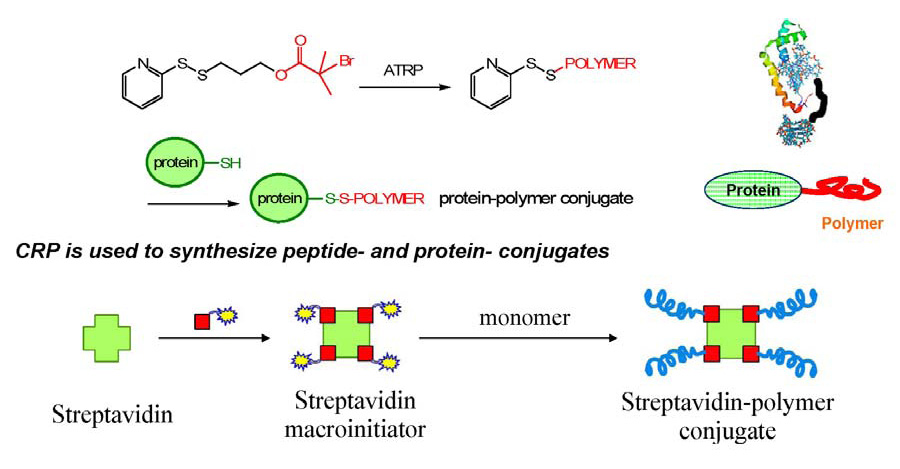
An example of a composite structure prepared by this route is the synthesis of near-uniform protein-polymer conjugates by initiating ATRP of monomethoxy poly(ethylene glycol)-methacrylate from 2-bromoisobutyramide derivatives of chymotrypsin, a protein-initiator. (12) However, initially the protein-polymer conjugates synthesized by this novel technique only retained 50-86% of the original enzyme activity and also suffered from challenging purification of intermediates and/or the inability to efficiently control the number or location of potential polymer connections, all of which can compromise the structural integrity of the modified protein.
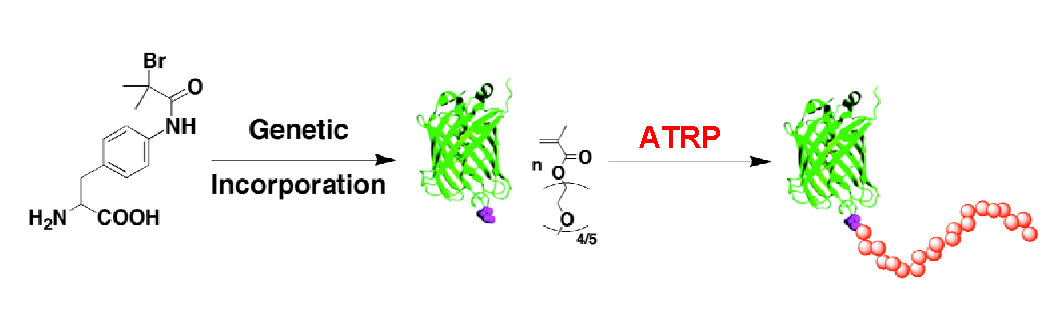
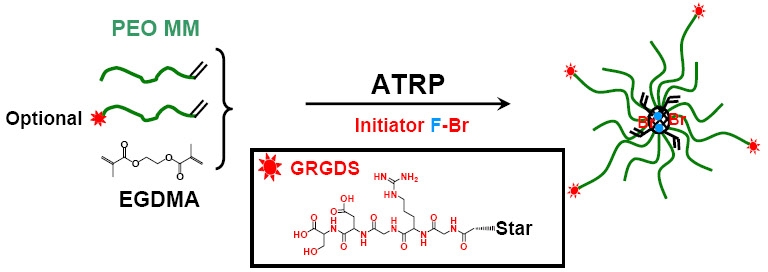
In order to address this deficiency and exemplify a general method for producing homogeneous recombinant proteins that contain a polymer initiator, or a functionality that can be converted into the desired functionality, at defined sites on the protein an amino acid containing a functional group that can act as an initiator for an ATRP was designed. The amino acid, 4-(2′-bromoisobutyramido)-phenylalanine, shown on the left hand side in the following scheme, can function as an initiator in ATRP and would provide a stable linkage between that particular amino acid, that was genetically incorporated into a specific site in the formed protein, and subsequent growing polymer chain from the linked initiator.
A green fluorescent protein was selected as the exemplary protein as any conformational change can result in the loss of fluorescence and so is a measure of the “severity” of the conditions employed for the ATRP on the functionalized protein. Polymerization initiated from the green fluorescent protein genetically modified at position 134 by incorporation of an amino acid modified to contain an ATRP initiator resulted in formation of a conjugate containing a single high MW polymer chain per protein molecule. (34)
The advantages of genetically engineering proteins are that they maintain the highest possible protein functionality, 95% with retention of tertiary structure in the case of GFP1, by protecting active sites and avoid structural weak regions and protection of immune active sequences while allowing precise control of initiator loading via amber codon. The resulting GFP1-poly(OEOMA) showed thermoresponsive behavior and retained activity.
This work was extended to allow an ARGET ATRP polymerization from a genetically encoded cleavable initiator to allow direct analysis of the grafted polymer after cleavage of the ester linkage in addition to providing enhanced initiation efficiency due to the faster initiation of secondary esters compared to corresponding amides. (35) The polymerization was conducted under biologically compatible conditions (36) using very low concentrations of catalyst and slow continuous addition of ascorbic acid in the presence of added halide salts to ensure the presence of a stable catalyst deactivator even at concentrations of catalyst as low as 300 ppm. The cleaved POEMA segments had Mw/Mn lower than 1.3. 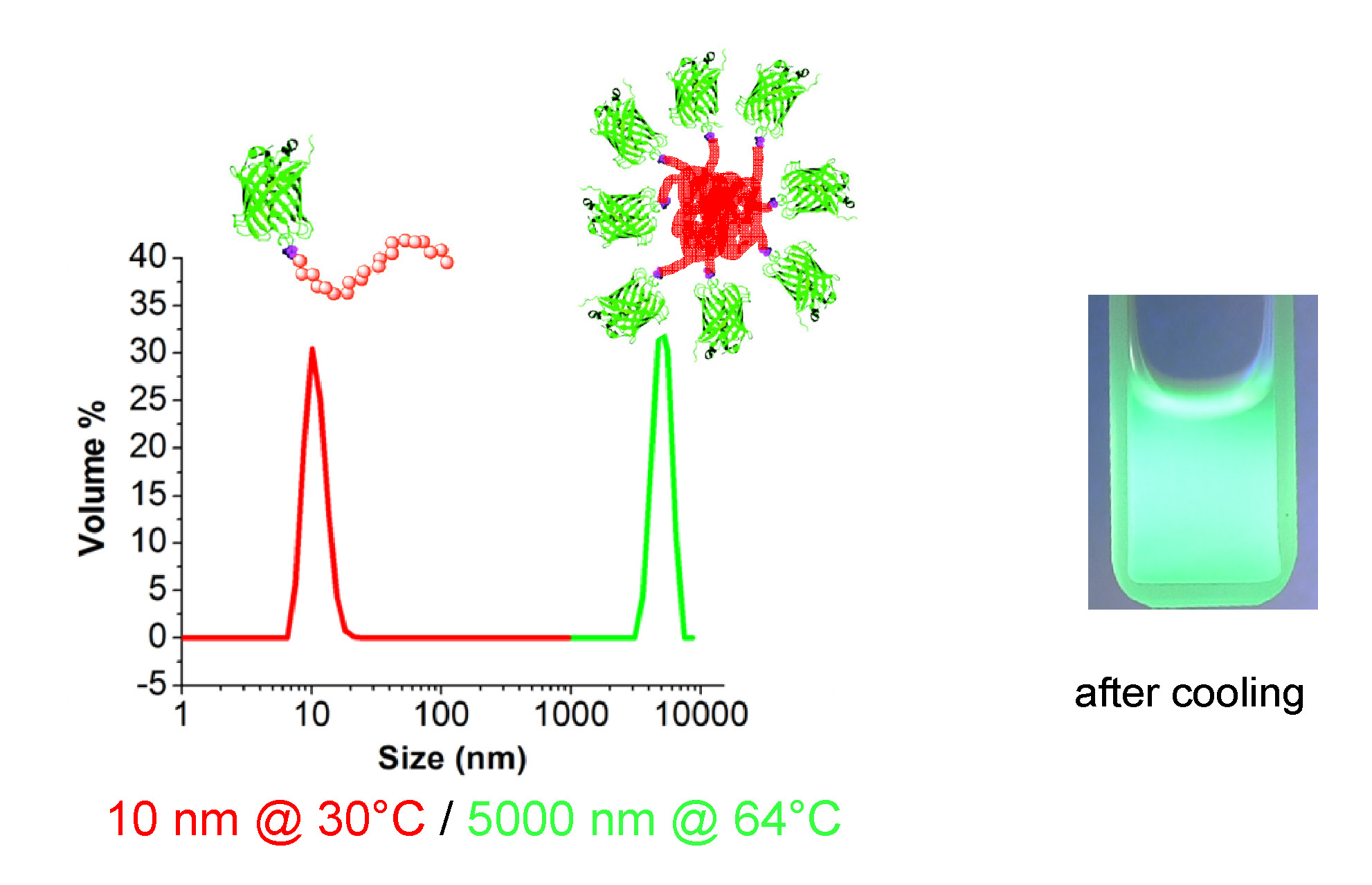
The protein conjugate could be incorporated into nanogels to assist in cellular delivery. This was accomplished by conducting a copolymerization of PEGMA and a PEG-dimethacrylate coinitiated by GFP1-initiator and a PEG-Br. The resulting nanogels provided a unique platform for protein delivery and enzymatic solid support systems. The successful incorporation of GFP1 demonstrates that proteins can be encapsulated whilst maintaining tertiary structure. Retained terminal functionality was used to attach catalase which converts hydrogen peroxide into water and oxygen thereby providing a simple assay for activity.
Composite structures prepared by this route include synthesis of near-uniform protein-polymer conjugates by initiating atom transfer radical polymerization of monomethoxy poly(ethylene glycol)-methacrylate from 2-bromoisobutyramide derivatives of chymotrypsin (a protein-initiator). (13) Protein-polymer conjugates synthesized by this novel technique retained 50-86% of the original enzyme activity.
Advantages of genetically engineering proteins are that one maintains highest possible protein functionality by protecting active sites thereby avoiding any structurally weak regions and protection of immune active sequences through precise control of initiator loading via amber codon.
Increasing the number of conjugated 2-bromoisobutyramide initiators per molecule of protein increased the MW and Mw/Mn of the final protein-polymer conjugates. The generic nature of this technique was demonstrated by initiating polymerization of nonionic, cationic, and anionic monomers from the protein-initiator. The technique described herein should be useful in synthesizing well-defined protein-polymer conjugates exhibiting a wide range of physical and chemical properties.
A significant extension of this procedure was the preparation of DNA block copolymers that are a unique class of biopolymer-synthetic polymer hybrids and have been used for targeted drug delivery and sensors.
Polymer-Nucleic Acid Bioconjugates
Nucleic acid (i.e. DNA and RNA) block copolymers (NABCp) are a distinct class of biomaterials that have applications in sensing and drug delivery. They can be synthesized using “blocking from” (BF) or “blocking to” (BT) methods. The term “blocking” is used as opposed to “grafting” to distinguish the growth of polymer from the chain-end, as in the case of nucleic acids, and growth from random points along a backbone, as in the case of proteins. This can be accomplished either in solution or directly from controlled porosity glass beads (CPG) solid phase. By tuning the properties of the DNA and the organic polymer, the properties of the DNABCp can be tailored. Such control has led to DNABCp’s that form reversible micelles and nanoscale assemblies, including some that are responsive to DNA-based recognition events.
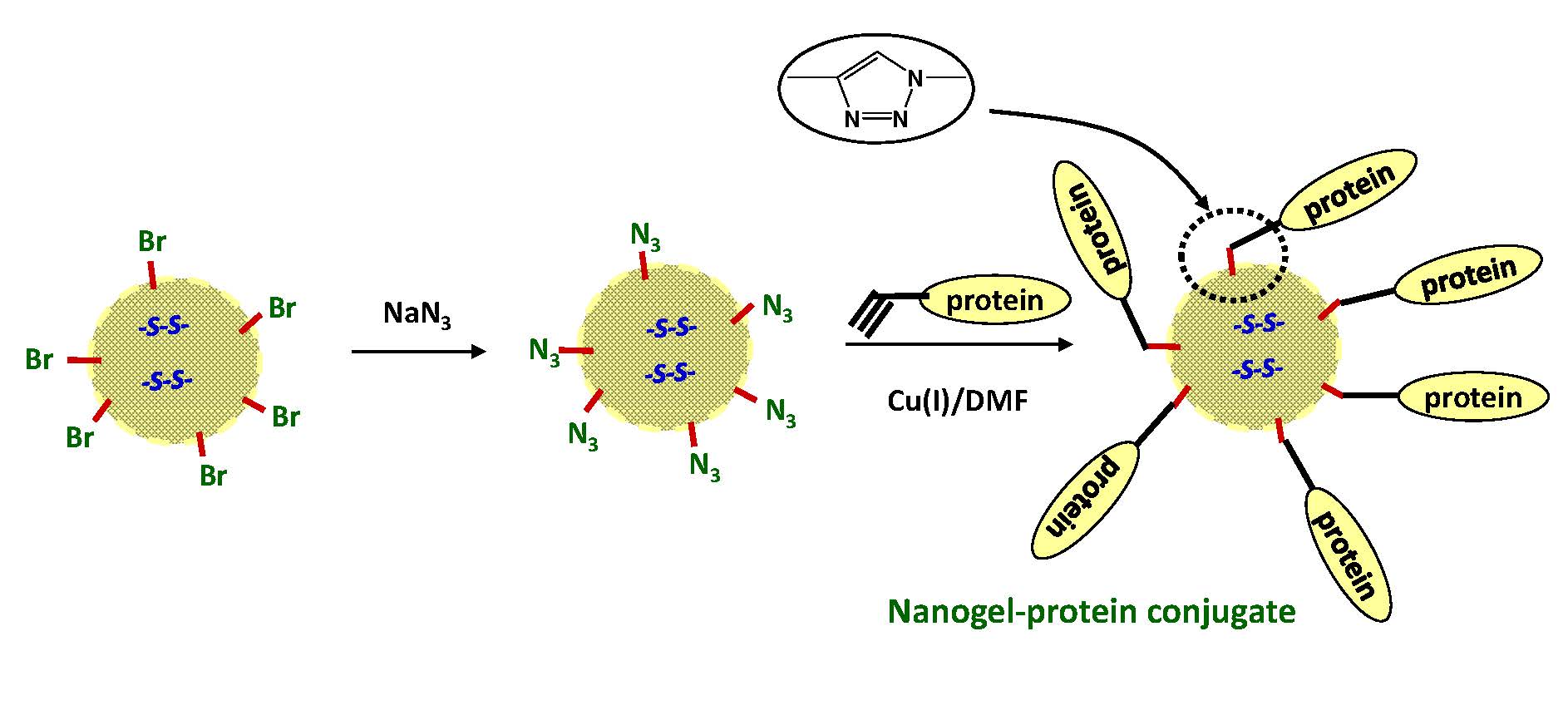
An early example of the blocking to procedure was a report discussing the preparation of star-polymer conjugates with DNA using a "ligandless" Cu(I) promoted azide-alkyne cyclo-addition click reaction. (44) The multivalency of the star-polymer architecture allows for the concomitant conjugation of other molecules along with the DNA, and the conjugation method provides control over the DNA orientation.
As illustrated below the star-polymer DNA nanoparticles were shown to assemble into higher-order nanoassemblies through hybridization. It was also shown that the DNA strands can be utilized in controlled disassembly of the nanostructures. The DNA star polymers were annealed to form a multiplexed assembly that could be disassembled using strand invasion of the hybrids.
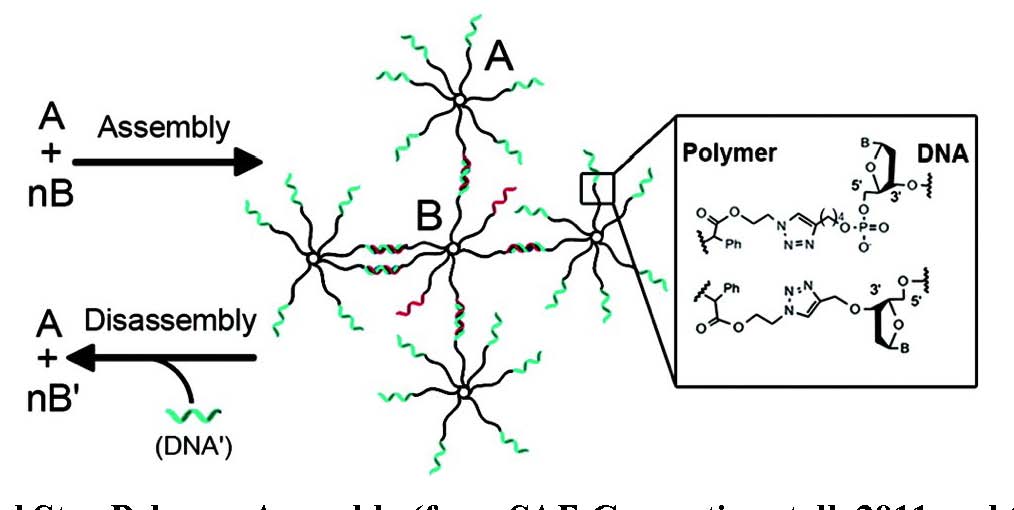
DNA Mediated Star Polymer Assembly (from SAE Consortium talk 2011 and (44)
There are two stringent requirements to prepare NABCp using solid phase methods: 1) the coupling reaction must have very high efficiency, and 2) the polymer formed in the GT reaction has to survive the relatively harsh NA deprotection protocol, i.e. concentrated ammonia for 4 h. The preferred coupling reaction to achieve high yields of NABCp is the use of phosphoramidite modified polymers. This requires the presence of a free hydroxyl group on the chain-end of the polymer that can be converted to a phosphoramidite thereby precluding the use of polymers with reactive side chains, i.e. hydroxyl, amine, carboxylic acid and azide groups among others. However, DNA block copolymers DNABCp were prepared by directly incorporating a phosphoramidite ATRP initiator into DNA during solid phase synthesis. (45) This method allowed for high yields and high purity of the DNA macroinitiators. Indeed the ability to use DNA-based supports expands the range of readily available molecules that can be used with the initiator, as exemplified by direct synthesis of a biotin polymer hybrid on a solid-support. This method expands the accessibility and range of advanced polymer biohybrid materials.
There are some advantages to this blocking from (b-f) strategy over the blocking to (b-t) approach. First, b-f allows for simpler purification of the low MW monomers and catalyst from the polymer bioconjugates. Second, in the b-f strategy the reactions are monomer addition reactions, whereas in b-t the conjugation reaction is between two incompatible large molecules (the polymer and the DNA), which is sterically more challenging.
Initially an ATRP initiator phosphoramidite capable of surviving typical DNA deprotection conditions (concentrated ammonia at room temperature for 4 h). An amide linked initiator was prepared from 4-aminobutanol and a-bromoisobutyryl bromide. An amide based initiator needed to prevent cleavage during DNA purification.
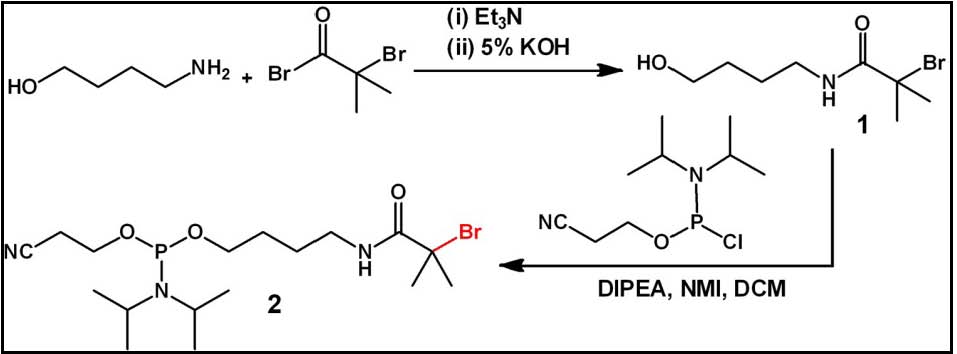
This ATRP initiator phosphoramidite could then be readily coupled to the 5’hydroxy group of a DNA sequence on a solid support, obtained after acid deprotection of the terminal dimethoxytrityl (DMT) group. The coupling of 2 to the terminal group was performed in 6 min using standard reagents in near quantitative yields. Thus, with this phosphoramidite coupling approach, an ATRP initiator could be directly incorporated into the DNA sequence during standard solid phase synthesis and afforded concomitant purification of DNA with terminal initiator functionality. This method offers both a simplified access and greater yield of the DNA with initiator. The monomer chosen for the polymer synthesis was oligo(ethylene oxide) methacrylate (OEOMA, Mn=475), owing to its biocompatibility and suitability in various biomedical applications. A small percentage of rhodamine methacrylate (RMA) was also incorporated into the reaction mixture to facilitate visualization and characterization of the resultant DNABCp.
Recently, procedures have been developed that allow ATRP under biologically relevant conditions using AGET ATRP. (36, 46, 47) The conditions for an AGET ATRP were optimized and used to prepare block copolymers of OEOMA or benzyl methacrylate, both in solution and from the solid phase bead. The DNA was shown to be stable during the blocking from polymerization.
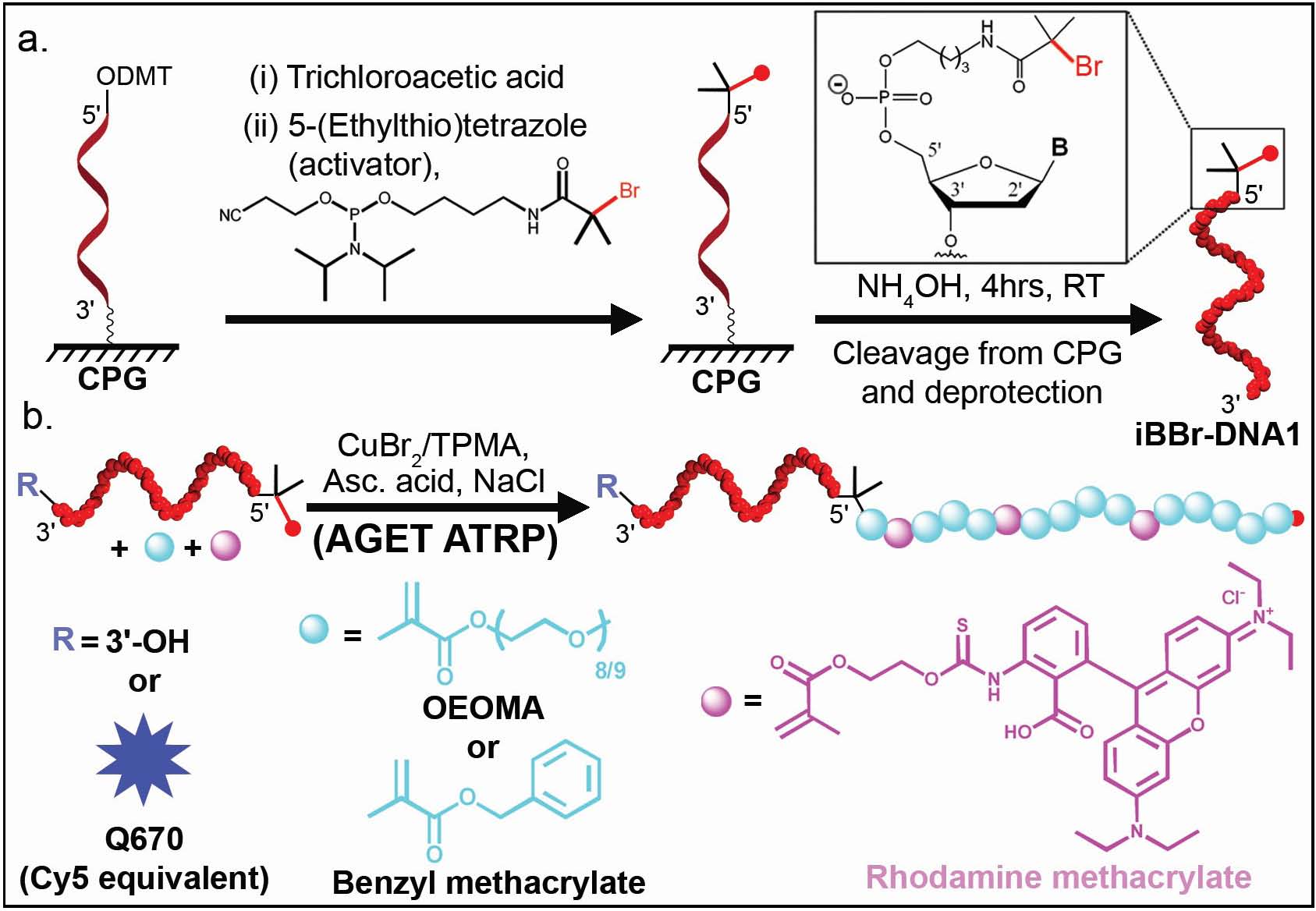
To determine if the polymerization affected the DNA structure, a second DNA-initiator sequence with a 3’terminal fluorescent dye, Quasar670 (Q670; a Cy5 equivalent) was prepared using similar conditions. If the polymerization caused cleavage of the DNA, there would be a loss of Q670 dye after purification of the reaction mixture. Conversely, if the polymerization did not lead to DNA scission, there would be Förster resonance energy transfer (FRET) from the rhodamine dye in the grafted polymer to the Q670 dye at the 3’-end of the DNA. The fluorescence spectra of the different hybrids show that FRET from the rhodamine to Q670 occurred only in the case of the Q670 terminated DNABCp but not when free dye was mixed with the DNA1-b-POEOMA-co-RMA. This indicated the DNA was not degraded during the polymerization.
DNA latex particles were prepared from DNA block benzyl methacrylate and could be used to selectively bind the DNA’s compliment strand, indicating potential of the DNA latex for DNA pull down experiments.
Polymers were also grown from the DNA initiator still attached to the solid support. It was thought that his method would facilitate not only the rapid preparation, but also the ready purification of functional bioconjugates from the unreacted monomers and catalyst.
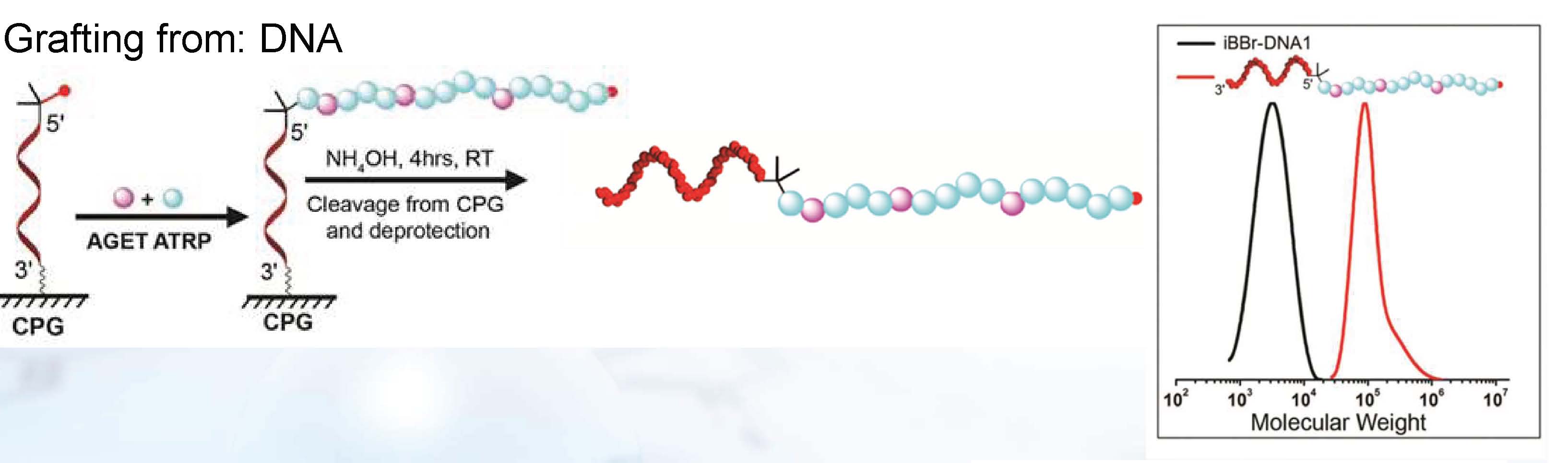
The solid phase initiator incorporation scheme was extended to ligate small molecule modified onto solid phase beads with initiators and graft polymer in situ thus providing a tool to prepare chain-end functionalized polymers in high purity.
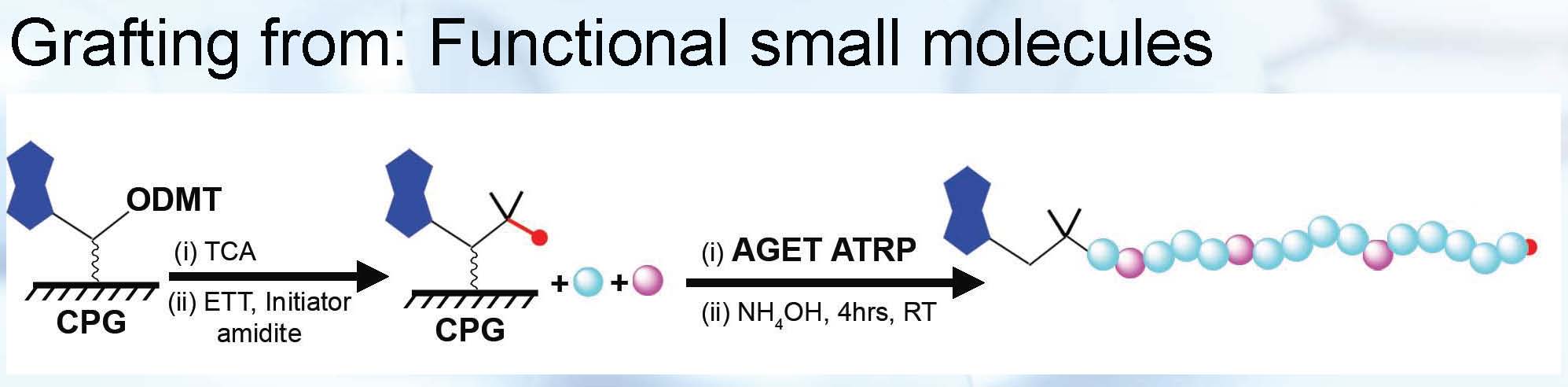
Another non-covalent approach involving preparation of delivery systems that does not require direct linkage of the biologically active agent to the synthetic polymer includes synthesis of cationically functionalized degradable star polymers for Short Interfering RNA, (siRNA) delivery. (36) This non-covalent bioconjugation approach is also based on formation of an ionic complex through electrostatic interactions. (35)
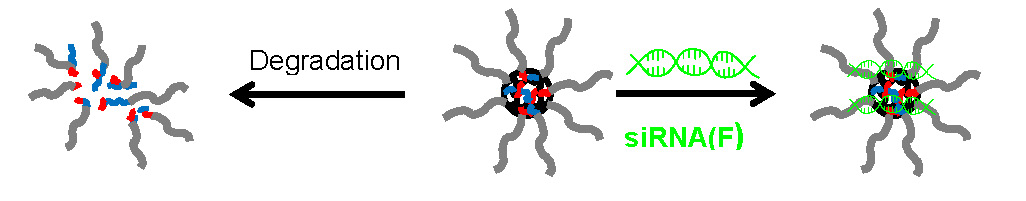
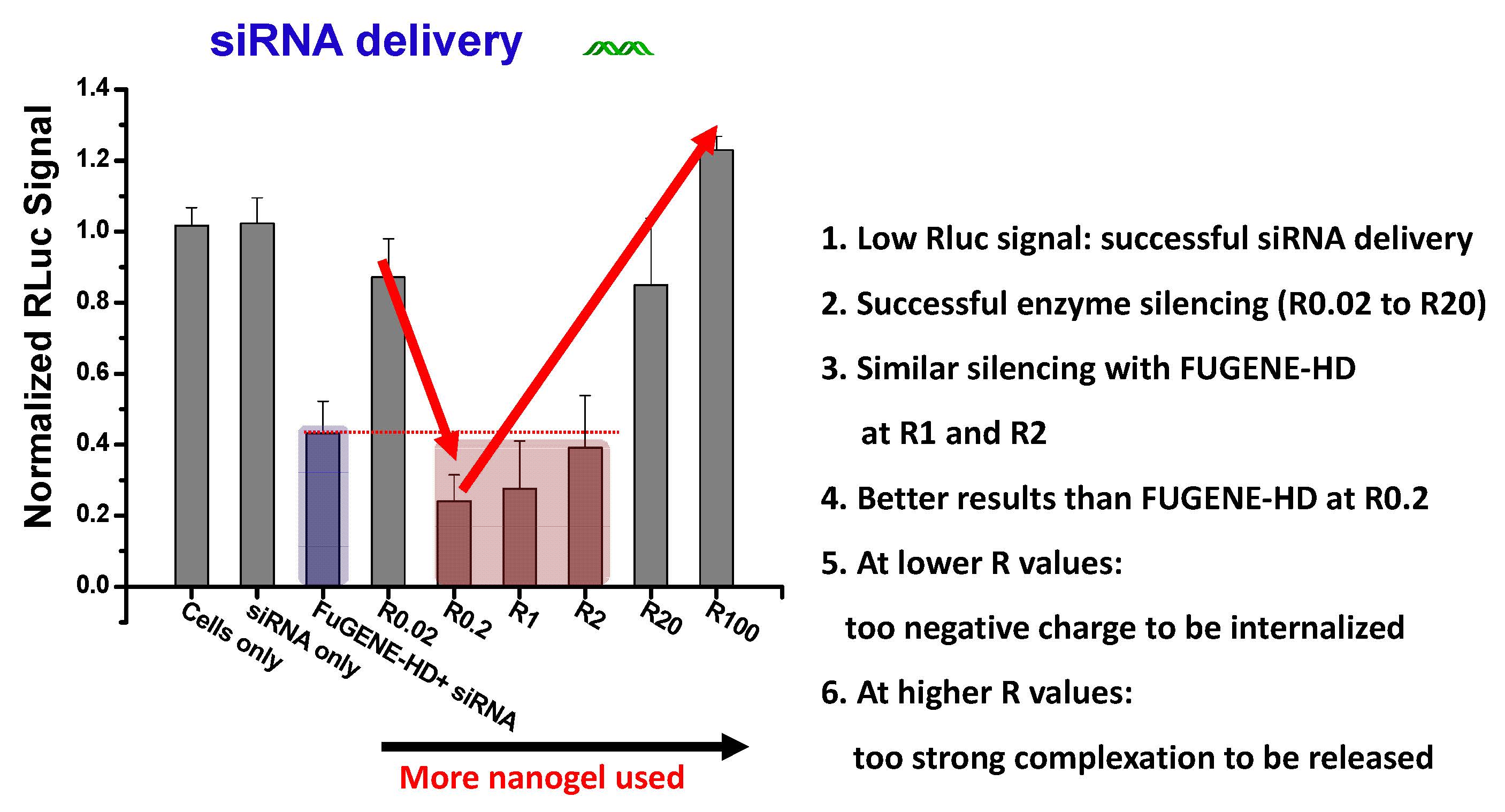
The requirements for delivery are that the bioconjugate forms a stable delivery system that allows prolonged circulation in the blood stream while incorporating functionality for targeted delivery through incorporation of specific ligands for receptors on target cells on the periphery of the first formed star. The size of the nanogels control cellular uptake and incorporation of biodegradable links allows for controlled release of the delivered biomolecules. (37) PEG based stars with cationic cores were evaluated for in vitro nucleic acid delivery. (38) The star polymers were combined with either plasmid DNA (pDNA) or short interfering RNA (siRNA) duplexes to form polyplexes for intracellular delivery. These polyplexes with either siRNA or pDNA were highly effective for NA delivery, particularly at relatively low star polymer weight or molar ratios, when tested with a dual luciferase reporter assay (Dual-Glo luciferase reporter assay, Promega) in Drosophila Schneider 2 (S2) cells highlighting the importance of NA release in efficient delivery systems. Most effective transfection results were obtained at lower ratios RW1, RW0.2, and RW0.02. They outperformed the current standard FuGENE-HD/siRNA conjugate. These results indicate that PEG-based star polymers with a cationic core can be utilized as efficient nonviral carriers for NA delivery, especially at relatively low RW or RM ratios.
Short Interfering RNA, (siRNA) was discovered in 1999. (39) siRNA is a double stranded RNA with 20-25 nucleotides with the negative charge due to the phosphate groups. There has been progress in developing cationic vectors for non-viral systemic gene therapy for treating hereditary diseases, but its potential role in the treatment of acquired diseases such as cancer is now widely recognized. (35) Dendrimers have been developed for siRNA delivery across the cell membrane (40) which would indicate that star macromolecules (41) which possess a similar core/shell structure with multiple arms, multiple surface functionality, and a globular topology should also work. The arm first route to star synthesis (42, 43) was employed to prepare PEG-star macromolecules a degradable core and incorporated peripheral functionality to tailor the stars for siRNA delivery to MC3T3-E1.4 cells. (44) Details of the synthesis were provided in the paper and successful degradation of PEG Stars using glutathione and under a physiological concentration of GSH demonstrated. siRNA formed a complex with the cationically charged DMAEMA/QDMAEMA core and confocal microscopy analysis showed complete cellular uptake in 24 hours. Another approach discussed was the preparation of N3-PEG Stars for siRNA delivery is being conducted in collaboration with Leroux’s group in Switzerland. The targeting agent is “clicked” to the star prior to siRNA complexation.
Surfaces for cell tissue engineering:
The final “star” topic is preparation of surfaces for cell tissue engineering. A series of photo-cross-linkable thermoresponsive poly(oligo ethylene oxide)methacrylate star polymers were designed to control of cell-surface interactions. The star macromolecules were prepared and deposited on a polystyrene surface. The star polymers covalently attached onto surfaces allowed a control of cell shrinkage and attachment in response to temperature changes. (45)
Biodegradable Nanogels:
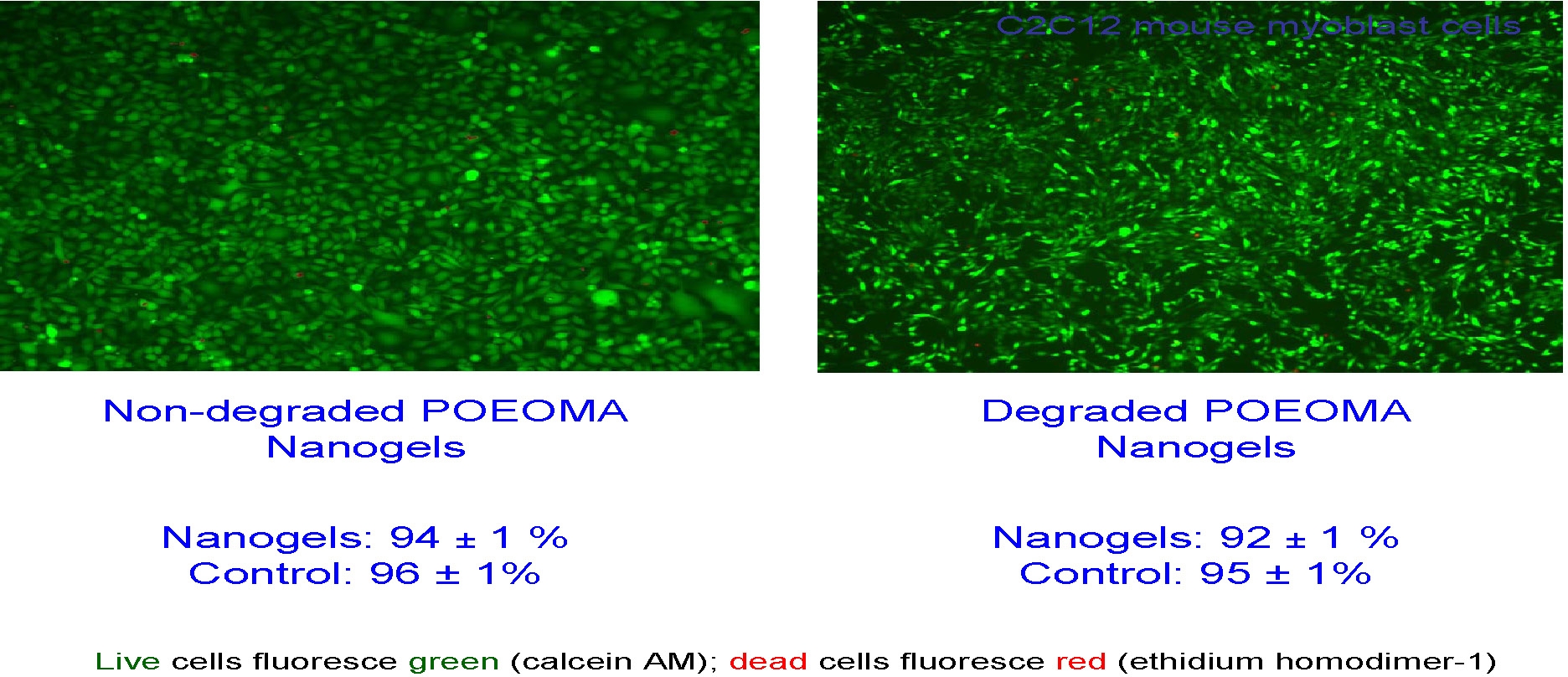
Stable biodegradable poly(oligo (ethylene oxide) monomethyl ether methacrylate), (POEOMA) nanogels, cross-linked with biodegradable linkages were prepared by conducting ATRP in a cyclohexane inverse miniemulsion copolymerization in the presence of a disulfide-functionalized dimethacrylate crosslinker. (46, 47) Biodegradable nanogels prepared by ATRP have several advantages over “classical” carriers for drug delivery applications since the networks are more uniform and degradation is more efficient. These nanogels could be used for targeted drug delivery scaffolds for biomedical applications since they can be degraded into lower molecular weight polymers to release the encapsulated (bio)molecules. These materials were proven to be nontoxic to cells and were biocompatible. (48)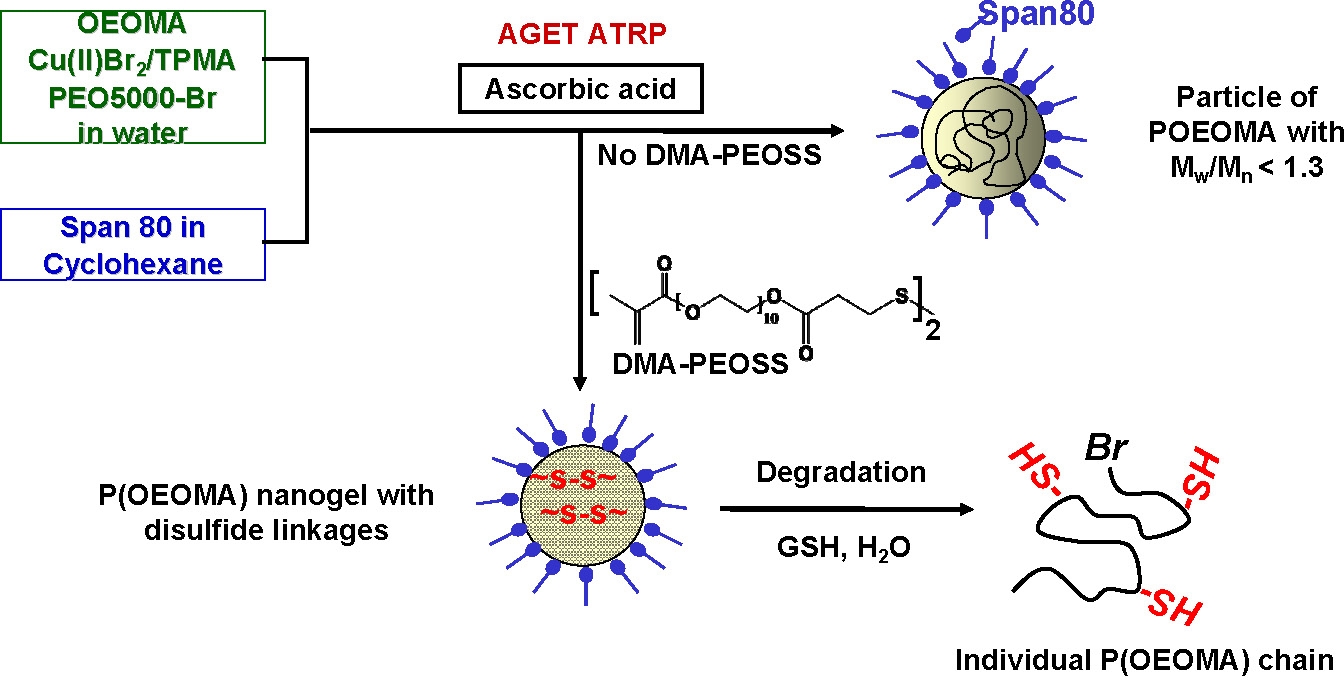
The nanogels consist of a uniform three-dimensional network, since they are prepared by a controlled polymerization process, and are capable of entrapping drugs, proteins, and nucleic acids and delivering them to specific targets. These nanogels can be degraded to individual polymeric chains using glutathione tripeptide, which is commonly found in cells at mM concentrations, as a reducing agent to release the encapsulated biomolecules. The encapsulation and controlled release of florescent dyes and Doxorubicin (Dox), an anticancer drug, was reported. (49) Results obtained from optical fluorescence microscope images and live/dead cytotoxicity assays of HeLa cancer cells suggested that the released Dox molecules penetrated cell membranes and therefore could suppress the growth of cancer cells.
Hydroxy-functionalized poly(oligo(ethylene oxide) monomethyl ether methacrylate) (HO-POEOMA) was prepared by AGET ATRP of OEOMA initiated by 2-hydroxyethyl 2-bromoisobutyrate in water, or in an inverse miniemulsion of water/cyclohexane, at ambient temperature to demonstrate facile applicability of the HO-POEOMA toward bioconjugation with biotin, pyrene, and GRGDS peptide. (50) The number of biotin molecules in each nanogel was determined to be 142,000 and the formation of bioconjugates of nanogels with avidin was confirmed using optical fluorescence microscopy. In addition, ATRP and click chemistry offered an efficient route for the synthesis of telechelic di-biotin polymers. These general methods can be applied to the formation of different functional materials conjugated with proteins, dyes, nucleic acids, and drugs.
ATRP allows the preparation of nano-gel materials with many useful features.
- First, the resulting particles preserve a high degree of halide end-functionality to enable further chain extension to form functional block copolymers or functionalization with biorelevant molecules by utilizing click reactions.
- Second, they are degradable in a reducing environment to individual polymeric chains with predeterminable molecular weight and relatively narrow molecular weight distribution (Mw/Mn < 1.5), indicating the formation of a uniformly cross-linked network in the individual particles. This uniform structure is anticipated to improve control over the release of encapsulated agents.
- Third, properties including swelling ratio, degradation behavior, and colloidal stability of particles prepared by ATRP are superior to those prepared by conventional free radical inverse miniemulsion polymerization.
- Fourth, nanogels will enhance circulation time in the blood, because they can consist of poly(oligo(ethylene oxide) monomethyl ether methacrylate) (POEOMA), an analogue of linear poly(ethylene oxide) (PEO) that can prevent nanoparticle uptake by reticuloendothelial system (RES).
These unique properties suggest that the well-defined functional nanogels prepared by this newly developed method hold great potential as useful biomaterials for biological and biomedical applications The procedure was also used to entrapped rhodamine isothiocyanate-dextran (RITC-Dx) as a model for water-soluble biomacromolecular drugs. (17) UV-Vis spectroscopy was used to characterize the extent of incorporation of RITC-Dx into the nanogels. The loading efficiency of RITC-Dx into the nanogels exceeded 80%. These nanogels were degraded into polymeric solutions in a reducing environment to release the encapsulated carbohydrate drugs. The released carbohydrate biomolecules specifically interacted with Concanavalin A in water, suggesting that the biodegradable nanogels could be used as carriers to deliver carbohydrate drugs that can be released upon degradation to bind to pathogens based on lectins.

The images above show a suspension of nanogels containing RITC-dextran in water before and after the addition of dithiothreitol (DTT) (a) and optical fluorescence microscopy images of nanogels loaded with RITC-dextran before (b) and after (c) degradation in water. The scale bars in b and c are 50 mm. These results demonstrate that biodegradation of nanogels can trigger the release of encapsulated molecules including RTIC-Dx and rhodamine 6G (fluorescent dyes), and Doxorubicin (Dox), an anticancer drug, as well as facilitate the removal of empty vesicles. A review was provided by the research group in conjunction with collaborators. (67)
Another example of the synthesis of biodegradable materials for a specific envisioned application was the preparation of poly(N-isopropylacrylamide-co-5,6-benzo-2-methylene-1,3-dioxepane) (poly(NIPAAm-co-BMDO)) copolymer which was synthesized by a combination of ATRP and reversible addition-fragmentation chain transfer (RAFT) polymerization. The lower critical solution temperature (LCST) of poly (NIPAAm) and poly(NIPAAm-co-BMDO) copolymers were measured using UV-vis spectroscopy to determine the effect of varying the amount of incorporated BMDO. (68) The material prepared is degradable and possesses a LCST above room temperature and below body temperature, making it a potential candidate for use as an injectable tissue engineering scaffold to enhance fracture repair. The combination of ATRP and RAFT enabled preparation of polymers with control over molecular weight, up to Mn = 50,000 g/mol and Mw/Mn < 1.2. Degradation studies were performed in basic solution and in complete Dulbecco's modified Eagle medium. The cytotoxicity of the material and its degradation products were analyzed by in vitro cell culture analyses, including cytotoxicity live/dead and CyQUANT cell proliferation assays. The materials consisted of crosslinked scaffolds with degradable units within the polymer backbone and at the crosslinking sites and were prepared using an ester-containing diacrylate crosslinker. Furthermore, incorporation of a GRGDS peptide sequence improved cell attachment to the gels providing further evidence that controlled/living radical polymerization techniques allow for precise control over macromolecular structure exemplified in this study by synthesis of materials suitable as a scaffold for tissue engineering.
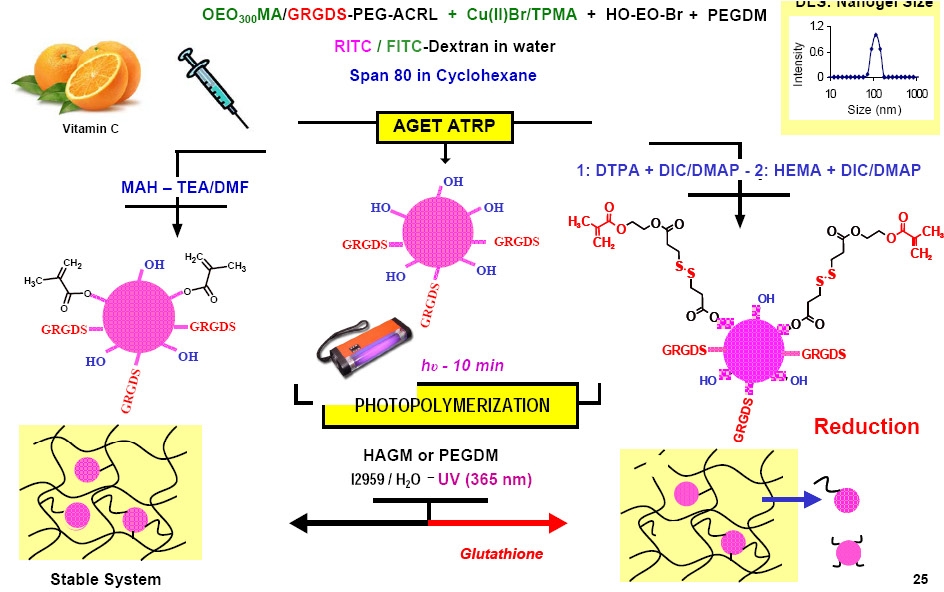
The influence of the degree of methacrylation on hyaluronic acid hydrogels properties was examined. (68) The properties of hyaluronic acid (HA) hydrogels having a broad range of methacrylation were determined and increasing solubility of glycidyl methacrylate (GM) in a co-solvent mixture during the methacrylation of HA with GM was shown to produce photopolymerizable HAGM conjugates with various degree of methacrylation (DM) ranging from 14% up to 90%. Aqueous solutions of HAGM macromonomers were photo-crosslinked to yield hydrogels with nearly complete vinyl group conversions after 10 min exposure under ultraviolet light (UV). The hydrogels were characterized by uniaxial compression and volumetric swelling measurements. Keeping the DM constant, the shear modulus was varied from 16 kPa up to 73 kPa by varying the macromonomer concentration. However, at a given macromonomer concentration while varying the degree of methacrylation of the hyaluronic acid (HA) hydrogels the shear modulus varied from 22 kPa up to 65 kPa. Preliminary in-vitro cell culture studies showed that GRGDS modified HAGM hydrogels promoted similar cell interaction at both low and high degrees of methacrylation, 32% and 60%, respectively. Densely cross-linked hydrogels with high degrees of methacrylation have been shown to be more mechanically robust while maintaining cytocompability and cell adhesion.
Extending this the work on hyaluronic acid hydrogels resulted in development of a new method to prepare nanostructured hybrid hydrogels by incorporating well-defined poly(oligo(ethylene oxide)monomethyl ether methacrylate) (POEO300MA) nanogels of sizes 110-120 nm into larger three-dimensional (3D) matrix forming hydrogels that are suitable for drug delivery scaffolds for tissue engineering applications. (69) Rhodamine B isothiocyanate-labeled dextran (RITC-Dx) or fluorescein isothiocyanate-labeled dextran (FITC-Dx)-loaded POEO300MA nanogels with pendant hydroxyl groups were prepared by activators generated electron transfer atom transfer radical polymerization (AGET ATRP) in a cyclohexane inverse miniemulsion. The resulting hydroxyl-containing nanogels were functionalized with methacrylated groups to generate photoreactive nanospheres.
1H NMR spectroscopy confirmed that polymerizable nanogels were successfully covalently incorporated into 3D hyaluronic acid-glycidyl methacrylate (HAGM) hydrogels after free radical photopolymerization. The introduction of disulfide moieties into the polymerizable groups resulted in a controlled release of nanogels from the cross-linked HAGM hydrogels in the presence of a reducing environment. The effect of gel hybridization on the macroscopic properties (swelling and mechanics) was studied and the results showed that swelling and nanogel content are independent of scaffold mechanics. In-vitro assays showed the nanostructured hybrid hydrogels were cytocompatible and the GRGDS (Gly-Arg-Gly-Asp-Ser) contained in the nanogel structure promoted cell-substrate interactions within 4 days of incubation.
These nanostructured hydrogels generate potential utility as an artificial extracellular matrix (ECM) impermeable to low molecular weight bio-active molecules with controlled pharmaceutical release capability. Moreover, the nanogels can provide controlled drug or bio-molecule delivery, while hyaluronic acid based-hydrogels can act as a macroscopic scaffold for tissue regeneration and regulator for nanogel release.
A further extension of biocompatible nanogels for bio-engineering applications was prepared by in situ entrapment of gold nanoparticles (AuNPs), bovine serum albumin (BSA), rhodamine B isothiocyanate-dextran (RITC-Dx), and fluoresceine isothiocyanate-dextran (FITC-Dx) using atom transfer radical polymerization (ATRP) in a cyclohexane inverse miniemulsion. (70) The nanogels consisted of poly(oligo(ethylene oxide) monomethyl ether methacrylate) (POEOMA), an analog of linear poly(ethylene oxide) which can prevent nanoparticle uptake by reticuloendothelial system that were covalently attached to a GRGDS peptide sequence for targeted delivery.
The nanogels were cross-linked using poly(ethylene oxide) dimethacrylate and initiated by a hydroxyl-functionalized initiator. Cell uptake of AuNP-loaded nanogels was verified using transmission electron microscopy (TEM). Cell internalization of BSA-loaded nanogels was confirmed using fluorescence microscopy after the addition of a fluorescently tagged antibody for BSA, and by running a western blot of the cell extracts. Confocal imaging of RITC-Dx-loaded nanogels demonstrated that the nanogels were internalized, and the location of the nanogels was confirmed by staining for actin and nuclei, as well as by following along the z-axis. Furthermore, co-localization of the RITC signal with a signal for an FITC-conjugated antibody for clathrin indicated that the nanogels enter via clathrin mediated endocytosis. The internalization of FITC-Dx-loaded nanogels with and without GRGDS into cells was quantified using flow cytometry and exceeded 95%.
These results confirmed that uniform nanogels prepared by ATRP in inverse miniemulsion are endocytosed and are applicable as drug delivery devices.
These results, and examples discussed in a more recent review article, (55) confirm that uniform nanogels prepared by ATRP in inverse miniemulsion are endocytosed and are applicable as drug delivery devices. Indeed the review article highlighted the fact that inverse miniemulsion ATRP was able to prepare stable sub-micron sized particles with diameter from 100 to 300 nm and narrow size distribution in the presence of a disulfide or polyester-functionalized crosslinker forming crosslinked biodegradable nanogels that were non-toxic when exposed to cells. The nanogel particles were capable of being loaded with various water soluble biomolecules and inorganic nanoparticles at high loading levels and high yield. Fluorescent dyes, AuNPs, and anticancer drugs were incorporated by physical loading, high molecular weight carbohydrates and proteins were incorporated by in-situ physical incorporation, and star branched polymeric nanoparticles by in-situ covalent incorporation. The nanogels crosslinked with polyester or disulfide linkages were biodegraded either in aqueous media or in the presence of biocompatible reducing agents such as glutathione, naturally present within cells. Upon degradation, encapsulated anticancer drugs such as Dox were released in a controlled manner to kill cancer cells. Carbohydrate drugs released from nanogels were bound to lectin such as ConA.
Since the nanogels preserved bromine chain end functionality this enabled further chain extension and selected modification and functionalization of the nanogels with biorelated molecules. Functional nanogels were also prepared by copolymerization with functional monomers or through the use of functional ATRP initiators. They were further functionalized with biorelated molecules, such as cell-adhesive peptides, targeting proteins and antibodies, resulting in the formation of nanogel bioconjugates. Finally, ATRP-nanogels had higher swelling ratios, better colloidal stability, and more controlled degradation than FRP-nanogels. These overall results suggest that well-defined functional nanogels may have potential as carriers for controlled drug delivery scaffolds to target specific cells.
Future design and development of effective microgel/nanogel based DDS utilizing CRP methods (not only ATRP) for in vivo applications to target cancer cells require a higher degree of control over properties. Targeted dimensions are in the range of 100 nm diameter, with a rapid environmental response for enhanced loading level, novel functionality for further bioconjugation, and biodegradability for sustained release of drugs. CRP in inverse microemulsion polymerization could produce nanogels with diameter less than 50 nm and with a uniform crosslinked network for better swelling. Appropriate selection of environmentally responsive water-soluble and water-swellable monomers will enable development of biomaterials responsive to external stimuli. In addition, introduction of acid-labile degradation linkages with/or without disulfide linkages will allow the DDS to be effectively degraded in the presence of targeted cancer cells.
Block Copolymers for Drug Delivery:
Acrylate-based block copolymers were prepared by ATRP as matrices for paclitaxel delivery from coronary stents. (56, 57) A series of multiblock acrylate-based copolymers, synthesized by ATRP were evaluated as drug delivery matrixes for the controlled release of paclitaxel from coronary stents. The copolymers consisted of poly(butyl acrylate) or poly(lauryl acrylate) soft blocks and hard blocks composed of poly(methyl methacrylate), poly(isobornyl acrylate), or poly(styrene) homo- or copolymers. Depending on the ratio of hard to soft blocks in the copolymers, coating formulations were produced that possessed variable elastomeric properties, resulting in stent coatings that maintained their integrity when assessed by SEM imaging of over-expanded stents. In vitro paclitaxel release kinetics from coronary stents coated with these copolymers typically showed an early release burst followed by sustained release behavior, which permitted the elution of the majority of the paclitaxel over a 10-day time period. It was determined that neither the nature of the polyacrylate (butyl or lauryl) nor that of the hard block appeared to affect the release kinetics of paclitaxel at a loading of 25% drug by weight, whereas some effects were observed at lower drug loading levels. Differential scanning calorimetry (DSC) analysis indicated that the paclitaxel was at least partially miscible with the poly(butyl acrylate) phase of those block copolymers.
The copolymers were also evaluated for sterilization stability by exposing both the copolymer alone and copolymer/paclitaxel coated stents to e-beam radiation at doses of 1-3 times the nominal dose used for medical device sterilization (25 kGy). It was found that the copolymers containing blocks bearing quaternary carbons within the polymer backbone were less stable to the radiation and showed a decrease in molecular weight as determined by GPC. Conversely, those block copolymers without quaternary carbons showed no significant change in molecular weight when exposed to 3 times the standard radiation dose. There was no significant change in drug release profile from any of the acrylate-based copolymers after exposure to 75 kGy of e-beam radiation, and this was attributed to the inherent radiation stability of the poly(butyl acrylate) center block.
A “greener” SARA ATRP catalyst was developed for the synthesis of PDMAEMA segmented copolymers by chain extension from cholesterol (CHO-Br) and PEG-Br. The iron0/CuII PMDETA based catalyst provided linear kinetics for the chain extension reactions with the PEG-Br initiator. The poor solubility of IPA CHO-Br in water was expected to lead to poor initiation efficiency but kinetics were linear generating bio-relevant block copolymers with narrow molecular weight distribution. (58)
Degradable Nanogels for Drug Delivery:
RDRP procedures have several inherent advantages for the preparation of functional materials for bio-related applications since in vivo applications require a high degree of control over the properties of the materials targeting specific environments. The requirements include particle stability for prolonged circulation in the blood stream, incorporation of peripheral functionality for further bioconjugation, controlled particle size with uniform diameter, and biodegradability for sustained release of drugs over a desired period of time and facile removal/degradation of empty devices. RDRP procedures such as ATRP provide a versatile route for preparation of (co)polymers with controlled molecular weight, narrow molecular weight distribution (i.e., Mw/Mn, or PDI < 1.5), designed architectures, and useful site specific chain end-functionality.
One of the initial steps towards preparing materials for bio-applications, within the Matyjaszewski group, started with incorporation of cross-linkers with degradable functionality, consisting of reversible redox cleavage of disulfide groups, into well-defined copolymers. (59) The evolution of the concept to resulted in formation of star macromolecules with bio-degradable cores (60) and bio-degradable gels (61) Such microgel/nanogel particles were evaluated as drug delivery carriers for biological and other biomedical applications. Disulfide linkages were selected as the degradable linkage to exploit tumor hypoxia in cancer treatment. (62)
The concept was further expanded by the evaluation of the utility of inverse miniemulsion ATRP to prepare well defined functionalized stable nanoparticles of water/swellable crosslinked polymers. The particles consisted of poly(oligo(ethylene glycol) monomethyl ether methacrylate) (POEOMA) containing a fraction, up to 100%, of degradable cross-linkages. (46) The use of ATRP for the formation of the degradable nanogels provided particles with higher degrees of swelling than materials prepared by standard free radical polymerization methods in both water, and organic solvents in addition to providing the ability to incorporate site selected functionality into the delivery vehicles. These nanogels could be used as targeted drug delivery scaffolds for biomedical applications, since the uniformly cross-linked network improved control over the rate of release of encapsulated agents. The nanogels underwent biodegradation forming water-soluble polymeric fragments, with a relatively narrow molecular weight distribution (Mw/Mn =1.5), and molecular weight below the renal threshold, confirming the formation of a uniformly crosslinked network in the individual particles.
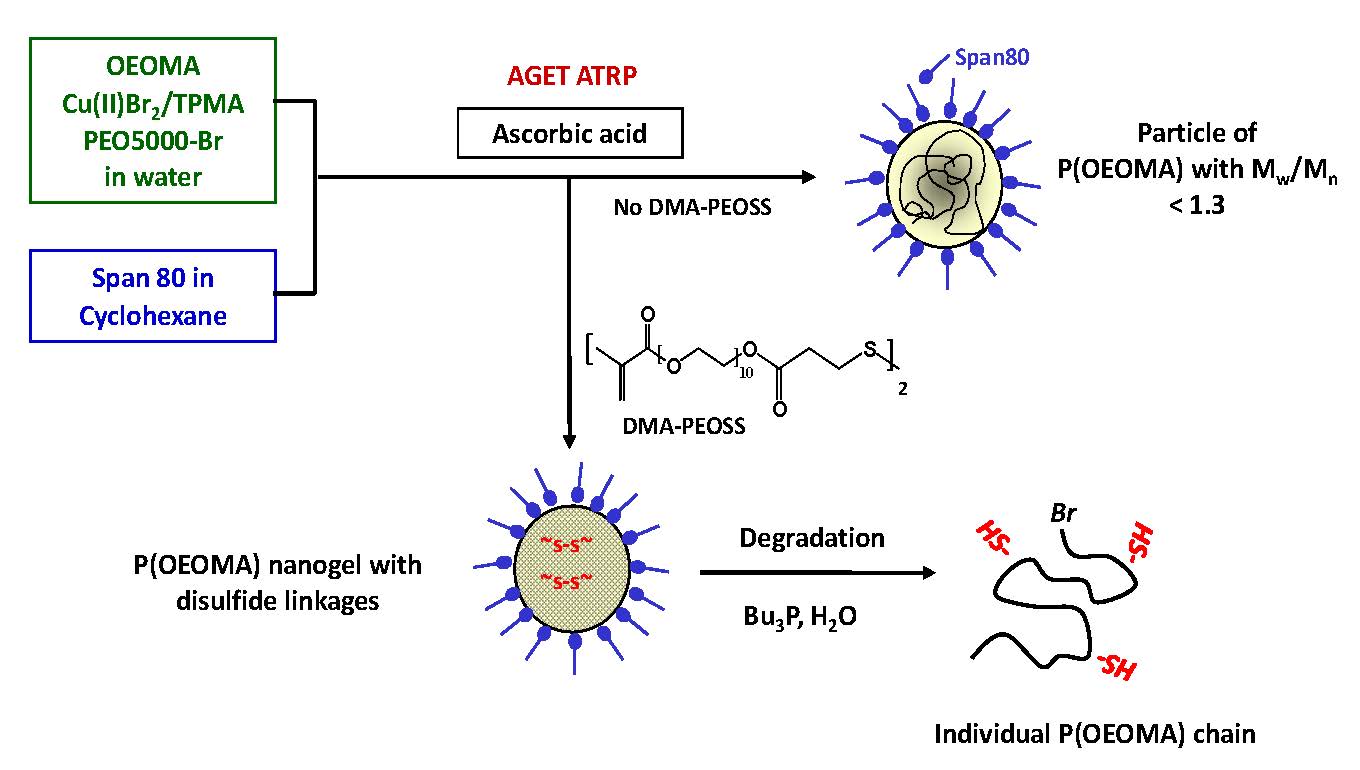
The uniform structure of the dual crosslinked particle was anticipated to improve control over the release of encapsulated agents in the presence of a biocompatible glutathione tripeptide, which is commonly found in cells. The biodegradation of the nanogels was exemplified by the triggered release of encapsulated molecules, including a fluorescent dye rhodamine 6G, and doxorubicin, an anticancer drug, as well as facilitating the removal of empty vehicles.
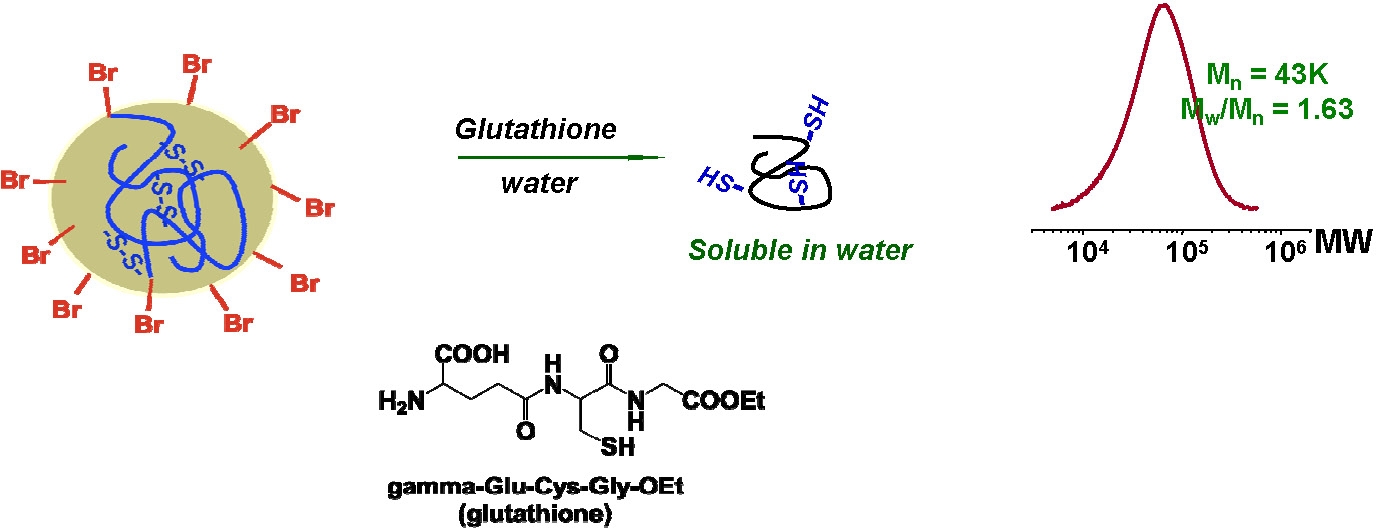
These nanogels can be degraded to individual polymeric chains using a reducing agent, including glutathione tripeptide, which is commonly found in cells at mM concentrations, to release the encapsulated biomolecules. These materials were proven to be nontoxic to cells and are biocompatible.
Biodegradable nanogels loaded with rhodamine B isothiocyanate-dextran (RITC-Dx) as a model for water-soluble biomacromolecular drugs were prepared using ATRP in a cyclohexane inverse miniemulsion in the presence of a disulfide-functionalized dimethacrylate cross linker. UV-vis spectroscopy was used to characterize the extent of incorporation of RITC-Dx into the nanogels. The loading efficiency of RITC-Dx into the nanogels exceeded 80%. These nanogels were degraded into polymeric sols in a reducing environment to release the encapsulated carbohydrate drugs. The released carbohydrate biomolecules specifically interacted with Con A in water, suggesting that the biodegradable nanogels could be used as carriers to deliver carbohydrate drugs that can be released upon degradation to bind to pathogens based on lectins. A similar procedure was used to load Dox and gold nanoparticles (AuNP) and loading was between 5.4.and16.4 wt% Dox depending upon initial ratio of Dox to nanogel and approximately 42% by mass of incorporated AuNP. (50, 54)
An extension of the work reported in earlier papers (18, 46) allowed the preparation of biomaterials with many useful features incorporated into the nanoparticles during the polymerization and using the incorporated site specific functionalities for post polymerization click functionalization providing a final product which can encapsulate bio-related molecules and/or inorganic nanoparticles such as pigments silica or quantum dots. The halogen functionality can be converted to another desired functionality, such as a group that will partake in a click conjugation reaction (63) while retaining the ability to be degraded in specific environments and release the encapsulated agents at a controlled rate. (17)
The procedures employed for this work have been summarized in reference (55) and a recent paper provided details on the cellular uptake of functional nanogels prepared by inverse miniemulsion ATRP with encapsulated proteins, carbohydrates, and gold nanoparticles and confirmed cellular uptake of nanogels was verified by transmission electron microscopy, gel electrophoresis, western blotting, confocal microscopy, and flow cytometry. (54)
The following figure shows the viability of HeLa cells after incubation for 48, 72, and 96 h. in three different systems that together show the validity of this approach to drug delivery, where the three colored bars represent:
(a) basic control without nanogels;
(b) Dox-loaded nanogels (0.4 mg/mL), where glutathione (0.08 mg/mL) was added after 48 h incubation to release Dox from Dox-loaded nanogels;
(c) control, where free Dox (0.064 mg/mL) was added after 48 h incubation.
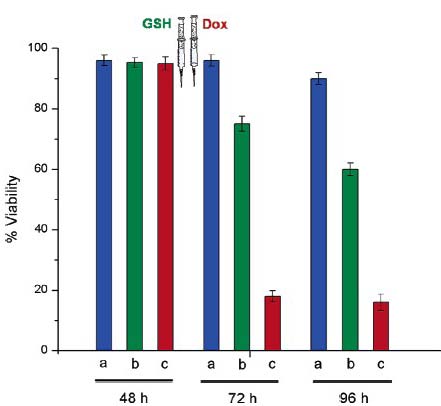
The figure shows that before addition of glutathione the viability of HeLa cells in the presence of Dox-loaded nanogels was similar to the control. This suggests that most of the encapsulated Dox remained in the nanogels with no significant non-specific leaching of Dox from the nanogels until after glutathione was added to the medium. After the addition of glutathione Dox was released at a slow controlled rate from the nanogels to kill the cancer cells.
This “passive” approach to encapsulate active agents for delivery to living tissues was expanded by preparation of nanogels and star macromolecules with incorporated cationic functionality for conjugation with nucleic acids. Cationic nanogels (qNGs ) were prepared for nucleic acid delivery. (80) The biodegradable qNG were prepared in a water-in-oil inverse miniemulsion utilizing AGET ATRP with CuBr2/tris(2-pyridylmethyl)amine (TPMA) as the ATRP catalytic species and poly(ethylene glycol) methyl ether 2-bromoisobutyrate (PEGMI2000, Mn = 2000) as a macroinitiator, for the copolymerization of oligo(ethylene oxide)methacrylate (OEOMA300, Mn = 300), and quarternized dimethylaminoethyl methacrylate (Q-DMAEMA) as comonomers, and dithiopropionyl poly(ethylene glycol) dimethacrylate (DMA) (Mn = 1260) as a crosslinking agent, and PEG2000OH as stabilizer.
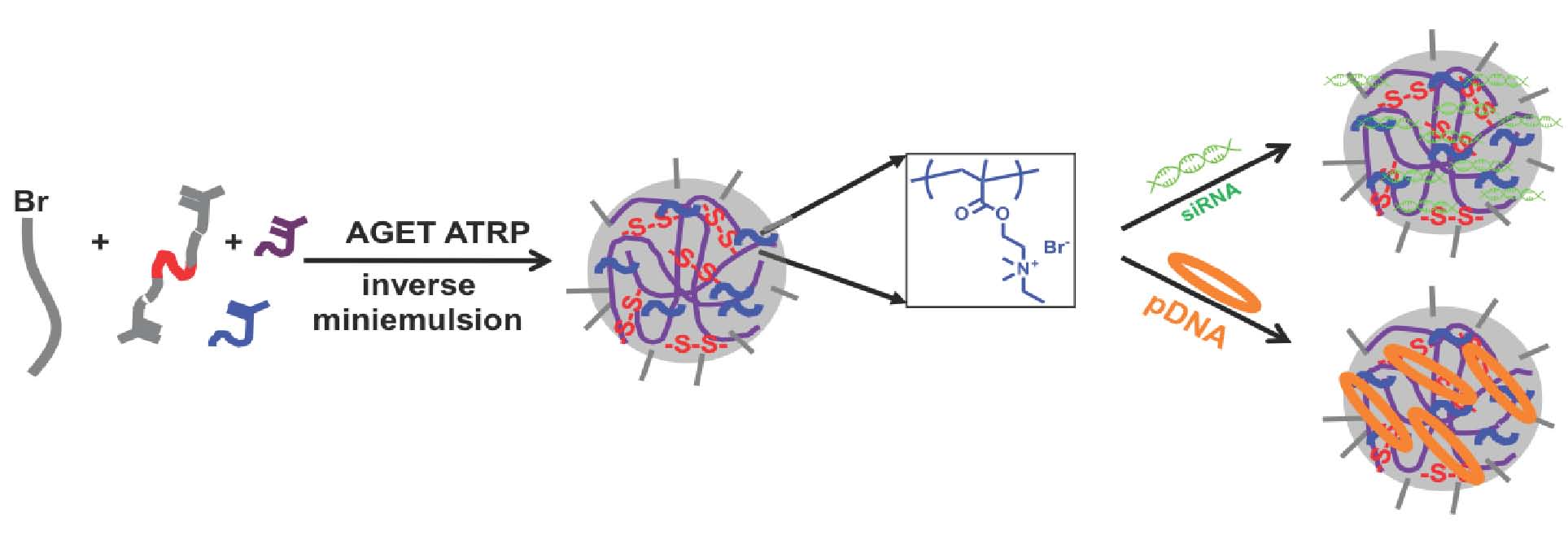
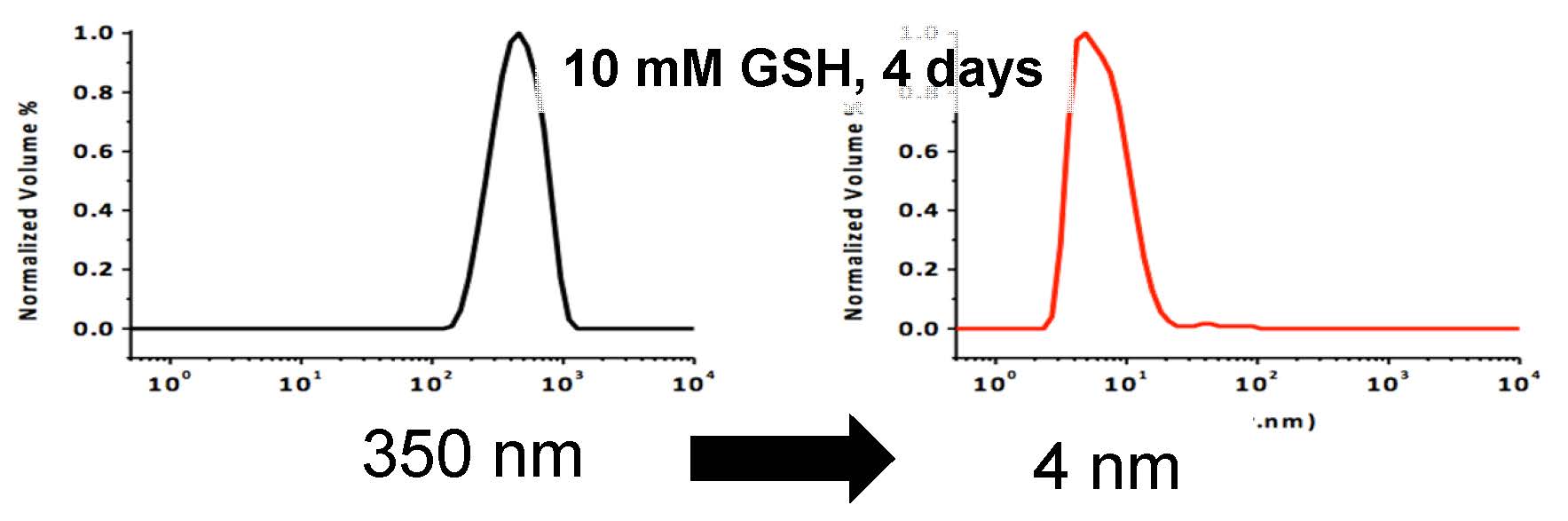
The qNGs were characterized by dynamic light scattering (DLS) and zeta potential, revealing a particle size of ca. 350 nm in diameter with a PDI of 0.164 and 17.9 mV +/-0.902, respectively. To determine the biodegradability of these qNG under reducing conditions, 1 mg/ml solutions of qNG in 10 mM glutathione were prepared and particle size was monitored using DLS. [10] The resulting degraded particles had a volume distribution of ca. 4 nm in diameter indicating a successful REDOX mediated degradation of the qNG.
Polyplex formation between the qNG and nucleic acid exemplified by plasmid DNA (pDNA) and short interfering RNA (siRNA duplexes) were prepared and evaluated.
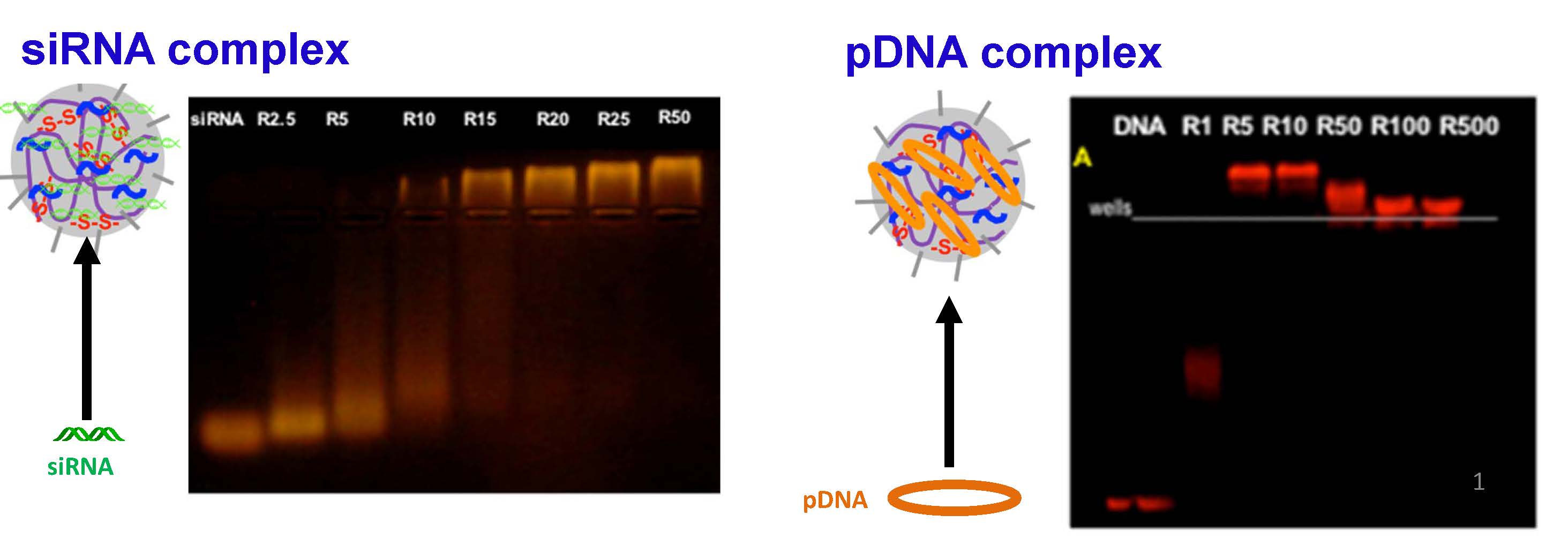
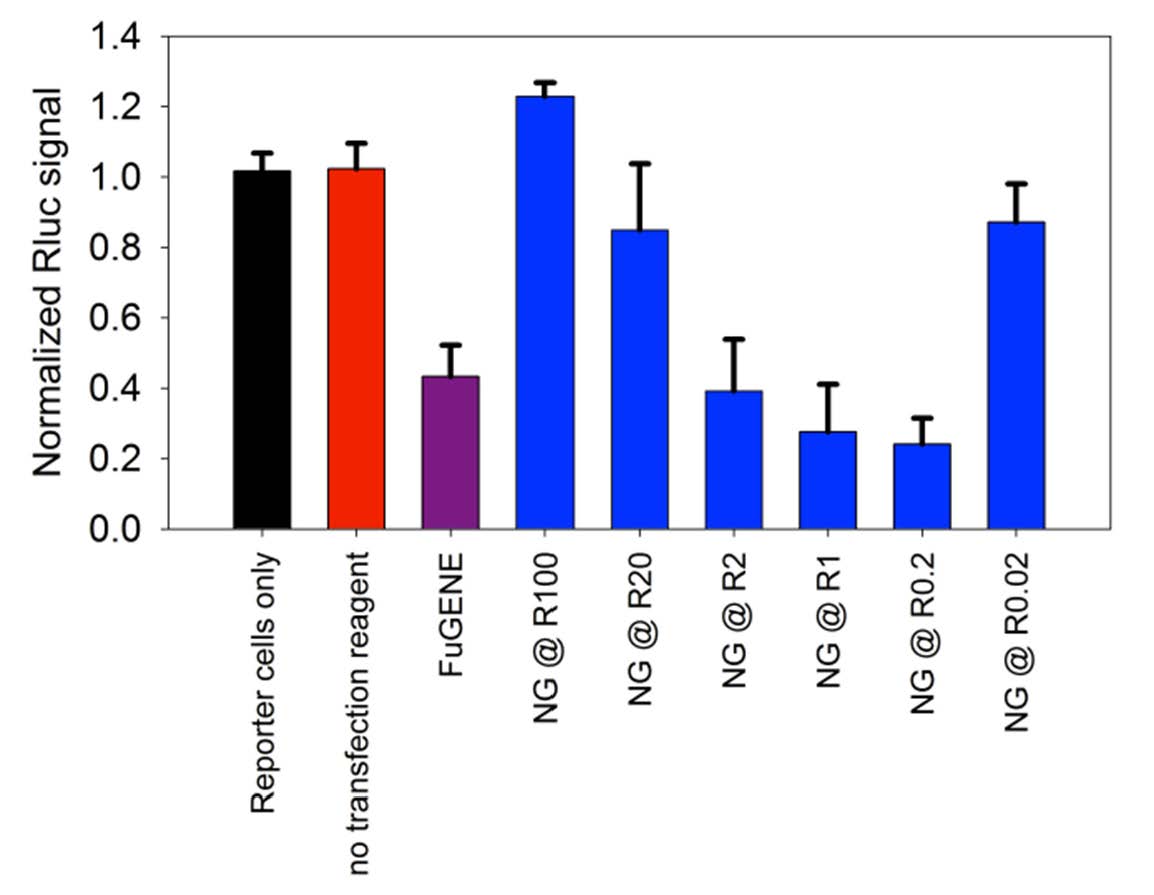
The delivery of the polyplexes was optimized for the delivery of pDNA and siRNA to the Drosophila Schneider 2 (S2) cell-line. The qNG/nucleic acid (i.e., siRNA and pDNA) polyplexes were found to be highly effective in their capabilities to deliver their respective payloads. The qNGs complexed pDNA and siRNA at relatively low weight ratios of qNG to DNA (R5) and qNG to siRNA (R15), respectively, as observed in agarose gel electrophoresi and provided a robust delivery system for nucleic acids, both plasmid DNA (~5 kb) as well as siRNA. In order to characterize the ability of different ratios of qNG to transfect siRNA, a Dual-luciferase reporter assay was utilized to rapidly and accurately screen knockdown efficiency. A maximum reporter knockdown was obtained at R0.2, suggesting more effective transfection than the current standard siRNA-Fugene-HD.
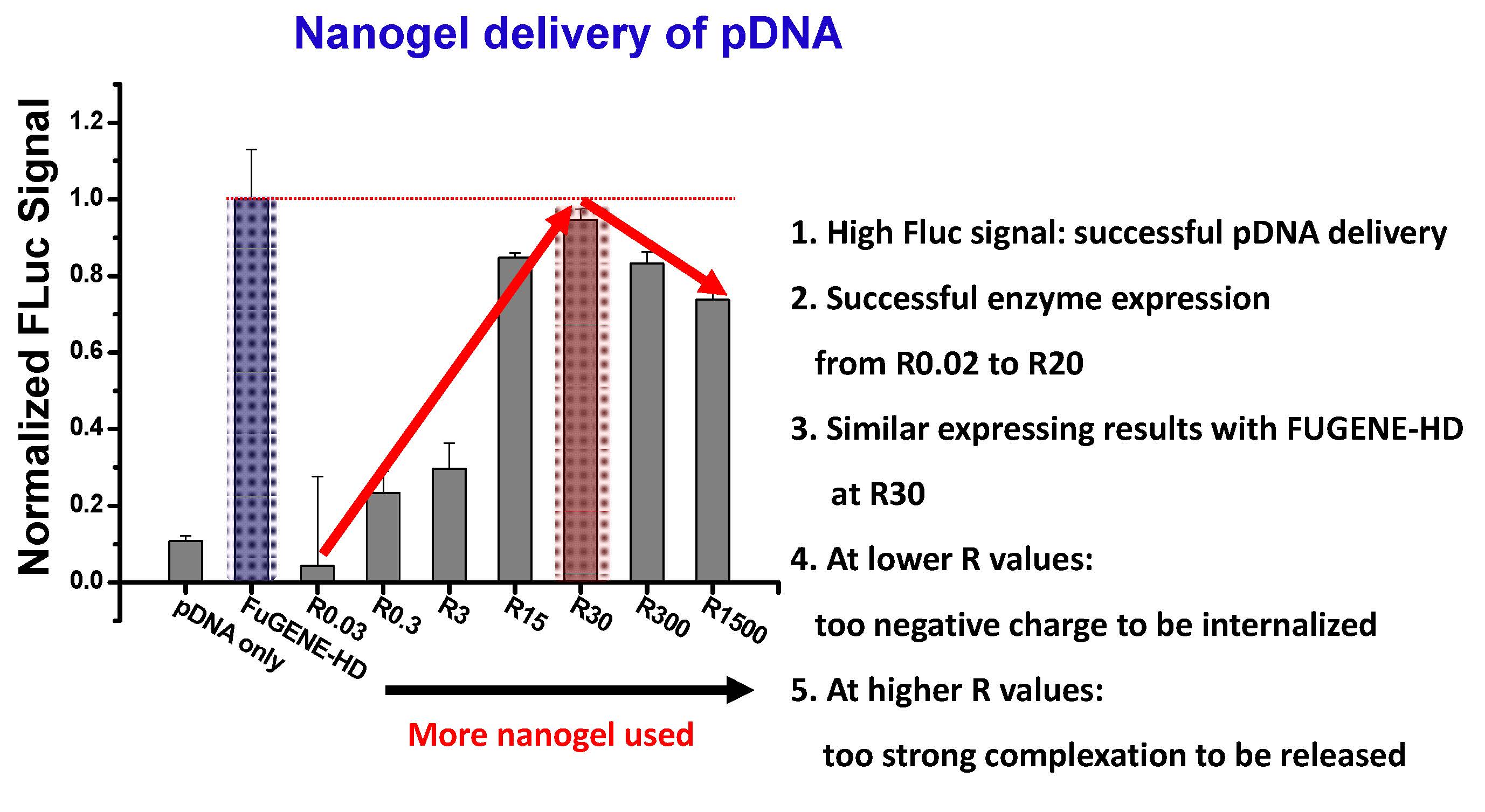
For plasmid DNA transfection, the maximum firefly luciferase reporter signal was observed at R30. These results confirm that qNGs are a promising platform for pDNA and siRNA delivery.
The use of magnetically directed nanoparticles for delivery of a drug is another application for bioactive nanoparticles. (81) A nanogel particle prepared by copolymerization of (diethyleneglycol) methyl ether methacrylate and a disulfide crosslinker in a microemulsion polymerization (82) was loaded with ferric oxide nanoparticles modified by oleic acid, size ~15 nm. The formed PM(EO)2MA displays a lower critical solution temperature (LCST) ~ 25oC. The magnetic nanoparticles were then loaded with ropivacaine to form a MNP/Ropiv complex.
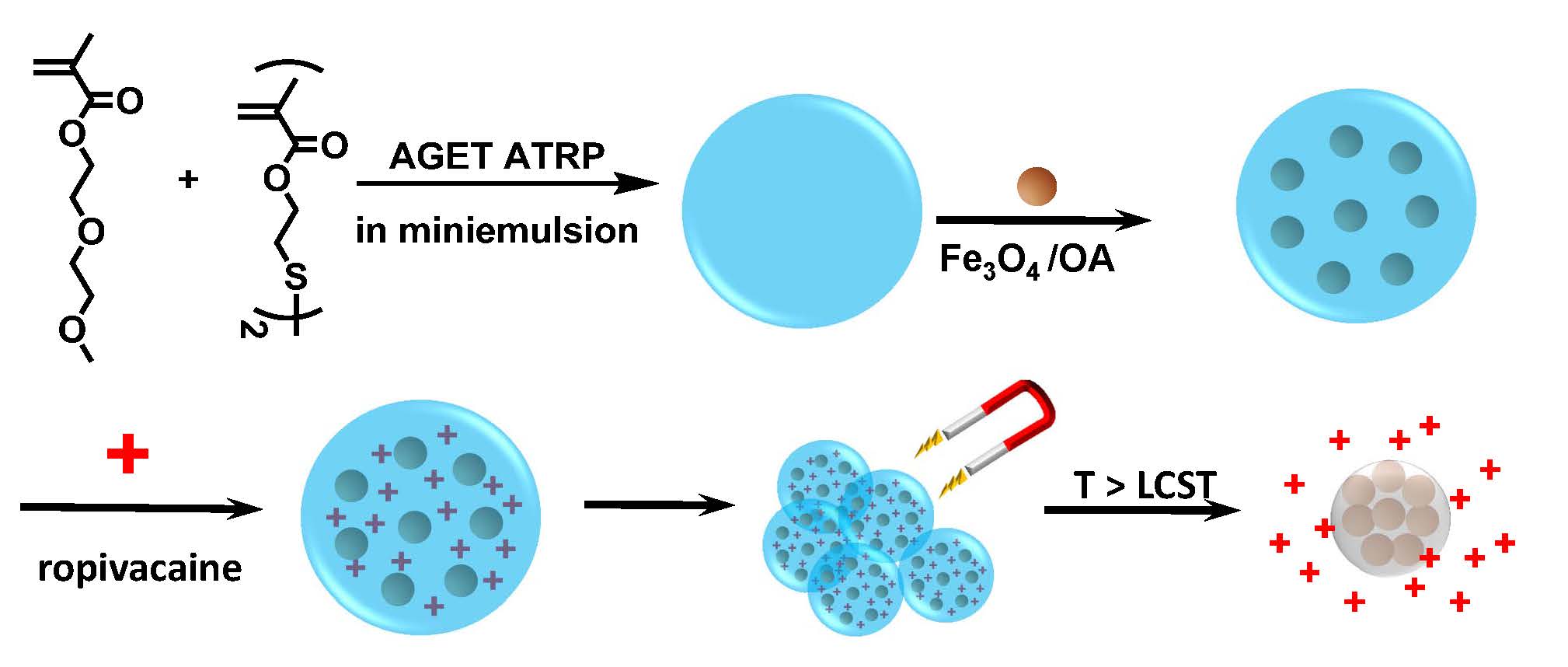
The MNP/Ropiv complex with magnetic application at the ankle of a rat resulted in irreversible release of the ropivacaine. The free drug acted on the nerves with which it came in contact. Delivery of a safe dose of ropivacaine via the MNP/Ropiv complex was 14 times higher than delivery by injection.
Another exemplification of the utility of nanostructured polymers for siRNA delivery was provided by a study on delivery targeting a mammalian cell line, murine calvarial preosteoblastlike cells embryonic day 1 subclone 4 (MC3T3-E1.4). Both stars and nanogels were tested for delivery and lower quantities of nanogels were needed to attain the same level of knockdown as the star polymers; there was no statistical difference between the knockdown efficiencies of the nanogel 5:1 and star 200:1 groups. The level of Gapdh knockdown in a mammalian cell is consistent with other cationic polymeric systems. The conclusion was that both star polymers and nanogels were biocompatible, and successfully complexed and delivered Gapdh siRNA, and suppressed Gapdh mRNA production to levels comparable to that of Lipofectamine® RNAiMAX. This underscores that the architecture of these NSPs do not compromise biocompatibility while maintaining knockdown efficiency.
Functional star molecules
As indicated above another approach to prepare functional materials for cellular internalization of transported agents was exemplified by the preparation of poly(ethylene glycol) (PEG) star polymers containing Gly-Arg-Gly-Asp-Ser (GRGDS) peptide sequences on the star periphery. The functionalized star molecules were synthesized by ATRP of poly(ethylene glycol) methyl ether methacrylate (PEGMA), GRGDS modified poly(ethylene glycol) acrylate (GRGDS-PEG-Acryl), fluorescein o-methacrylate (FMA) macromonomer, and ethylene glycol dimethacrylate (EGDMA) via an “arm-first” method.
The star polymers were approximately 20 nm in diameter, as measured by dynamic light scattering and atomic force microscopy. Conjugation of FMA to the stars was confirmed by fluorescence microscopy, and successful attachment of GRGDS segments to the star periphery was confirmed by 1H NMR spectroscopy. Both fluorescent PEG star polymers with and without peripheral GRGDS peptide segments were cultured with MC3T3-E1.4 cells. These star polymers were biocompatible with greater than 90% cell viability after 24 h of incubation. Cellular uptake of PEG star polymers in MC3T3-E1.4 cells was observed by confocal microscopy. Rapid uptake of PEG star polymers with GRGDS peptides (∼100% of FITC-positive cells in 15 min measured by flow cytometry) was observed, suggesting enhanced delivery potential of these functional star polymers. (60)
The concept behind using stars for delivery applications was that the size of the core of the star could be adjusted to provide an appropriate degree of complexation that allowed efficient siRNA complexation but also sufficiently weak complexation that allowed release. This balance could be attained by adjusting the degree of protonation in the core. (52, 60) The conditions for the preparation of the star copolymer that provided the best result is shown below.
|
|
Mw, THF GPC(×103) |
Mw/Mn |
conv,DMAEMA |
conv,EGDMA |
Mw, MALLS(×103) |
Arm/star |
N/star |
Dh (nm) |
|
|
31.2 |
1.33 |
81 % |
87 % |
120.8 |
48 |
79 |
13.3 |
There was complexation between the core of the star and siRNA at low star to siRNA ratios as measured by zeta potential and a star ratio to siRNA of 0.02 to 0.2 provided better enzyme silencing than FuGENE while DNA star delivery for enzyme expression was improved, with a performance better than FUGENE-HD, at ratio of star to DNA of 15.
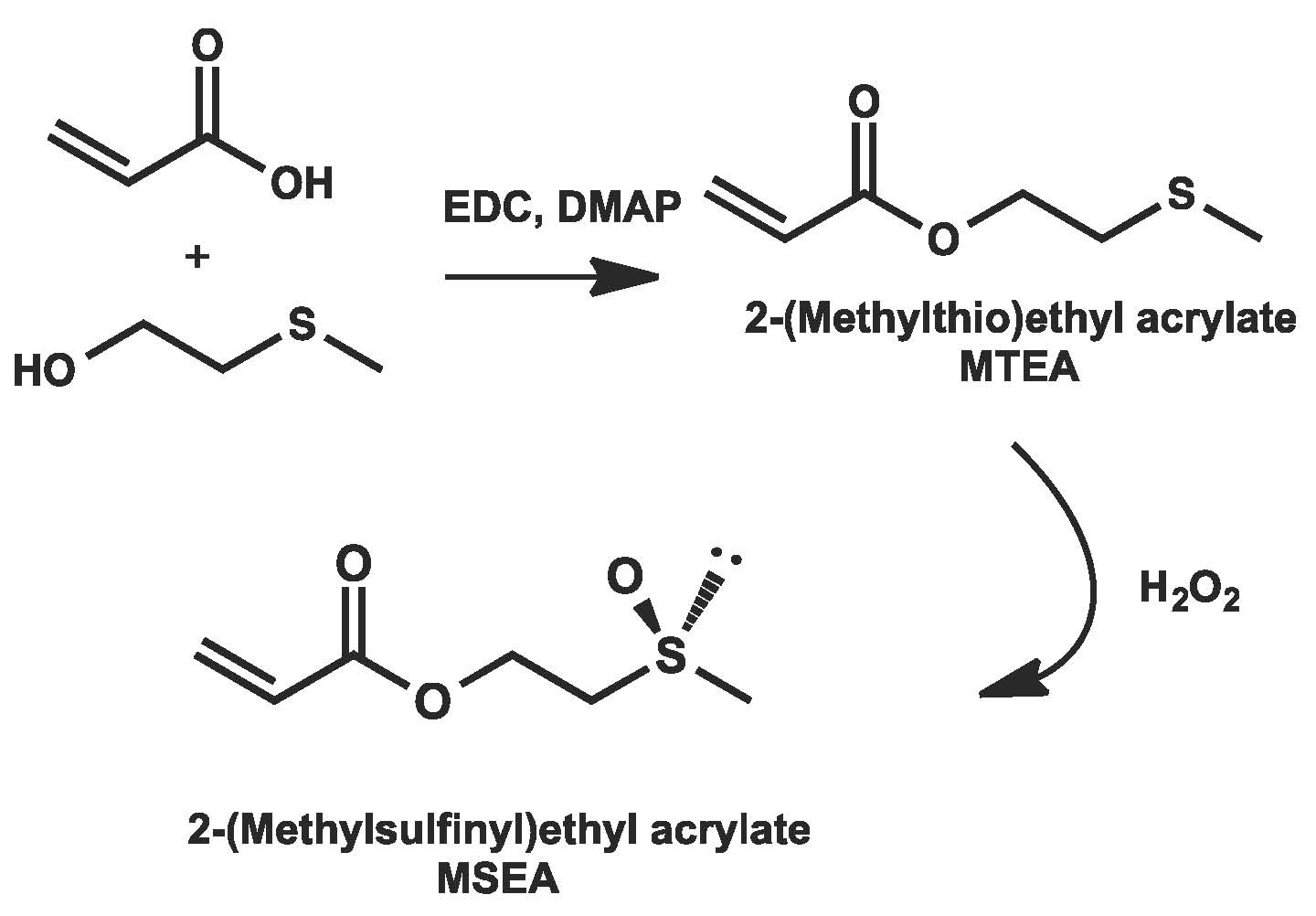
PEG is among the most widely used polymers in biomedical applications due to its hydrophilicity, water solubility, and chemical stability of the ether groups. Its neutral charge prevents interactions with negatively charged cell membranes, or opsonins that enhance phagocytosis and polymer clearance and PEGylated species can circulate longer in the bloodstream. However, the conjugation of monofunctional PEG to biorelevant molecules may result in the formation of heterogeneous products due to the presence of bifunctional PEG impurities and low molecular weight PEG may aggregate at elevated temperatures due to its lower critical solution temperature (LCST). In addition, it has been recently reported that the immunogenicity of PEG may result in inefficient drug delivery and severe immune reactions. (83) In order to overcome the limitations of PEG based conjugates a new type of biocompatible polymer was developed: a polymeric analog of dimethyl sulfoxide (DMSO) namely poly(2-(methylsulfinyl)ethyl acrylate) (polyMSEA). DMSO is a neutral uncharged molecule used as a transdermal delivery enhancer, polymerase chain reaction inhibitor, cryoprotectant, and in veterinary medicine. The biocompatibility of DMSO is largely due to the presence of the polar aprotic methyl sulfoxide group. (84)
Inspired by these unique properties of DMSO, we aimed to synthesize a polymeric analogue of DMSO by ATRP with high water-solubility and low cytotoxicity. (85) 2-(Methylthio)ethyl acrylate (MTAE) was prepared, as shown in the following schematic, and it could be directly polymerized and then oxidized to the PDMSO analog or converted to 2-(methylsulfinyl)ethyl acrylate prior to polymerization.
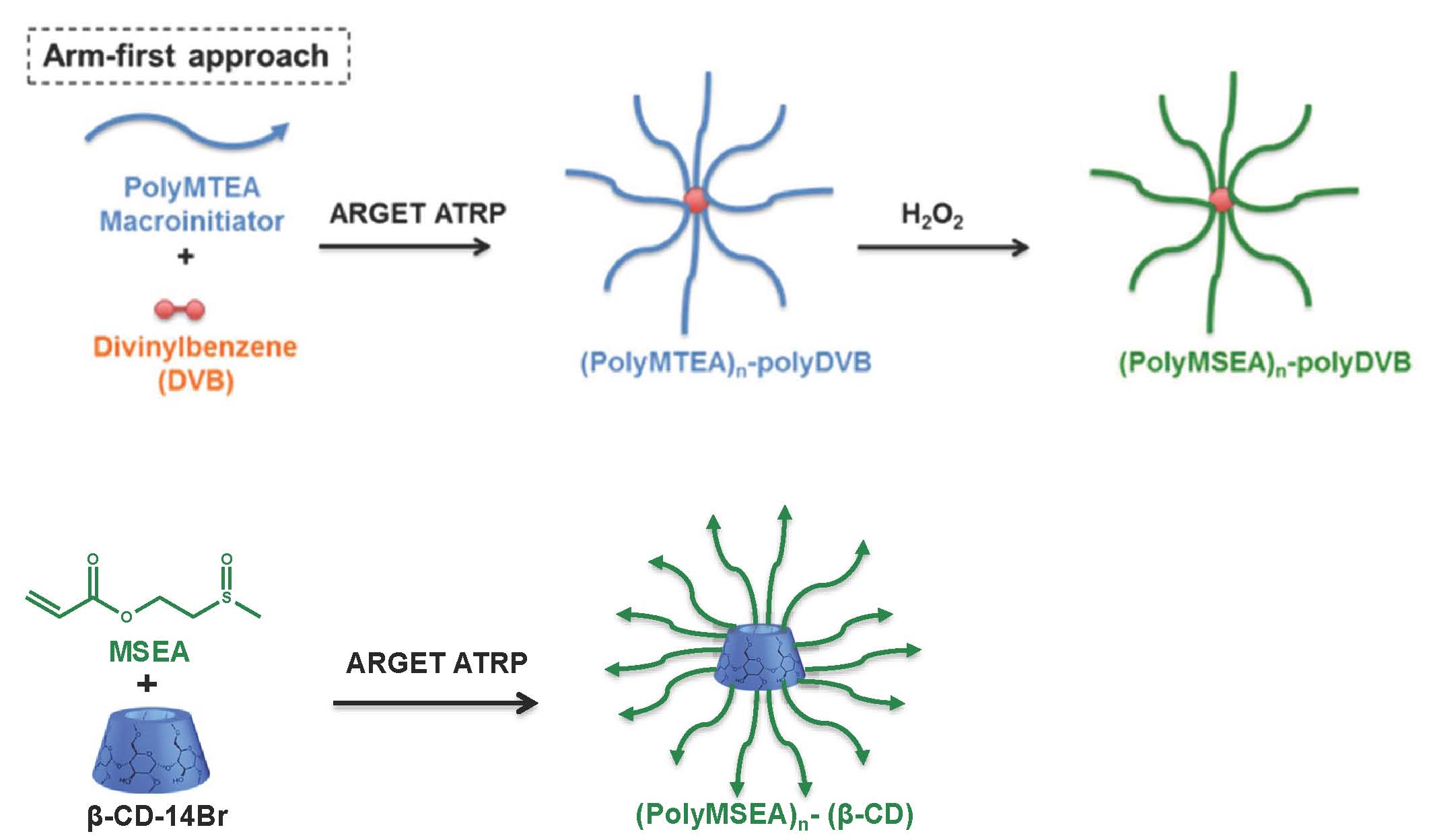
Well-defined linear polyMSEA with low dispersity (<1.3) was synthesized by ARGET ATRP in the presence of 100 ppm copper catalyst. In addition, two well-defined star polymers were prepared, one using a “core-first” approach with a biodegradable β-cyclodextrin core and the other one by an “arm-first” approach with divinylbenzene as cross-linker.
The LCST of pure polyMSEA (ca. 140 °C) was estimated from the solution aggregation points of copolymers of MSEA and NIPAM with different compositions. The cytotoxicity of neutral water-soluble linear and star-shaped polyMSEAs was evaluated using human embryonic kidney cells (HEK 293). Both linear polyMSEA and (polyMSEA)n-polyDVB star showed no toxicity, even at concentration up to 3 mg/mL in comparison to the control group (only cell without addition of other chemicals). This result proved that polymeric analogues of DMSO prepared by ATRP possess low cytotoxicity, similar to their monomeric counterpart: DMSO.
Therefore one can say that these polymeric analogues of DMSO have the following advantages: (1) straightforward synthesis from commercially available starting materials,
(2) low degrees of steric hindrance, high water-solubility and low cytotoxicity, and
(3) they possess the potential of bio-conjugation by grafting-onto or grafting-from (bio)-functional compounds/substrates via ATRP.
Therefore the water-soluble and nontoxic polymeric analogue of DMSO could be used in some biomedical applications and/or potentially replace such polymers as PEG, polyOEGMA, and polyoxazolines in bio-conjugates. Indeed these new polymers could potentially be considered as an alternative to PEGylation approach in some biomedical applications.
Biolubrication (wear protection without surface modification)
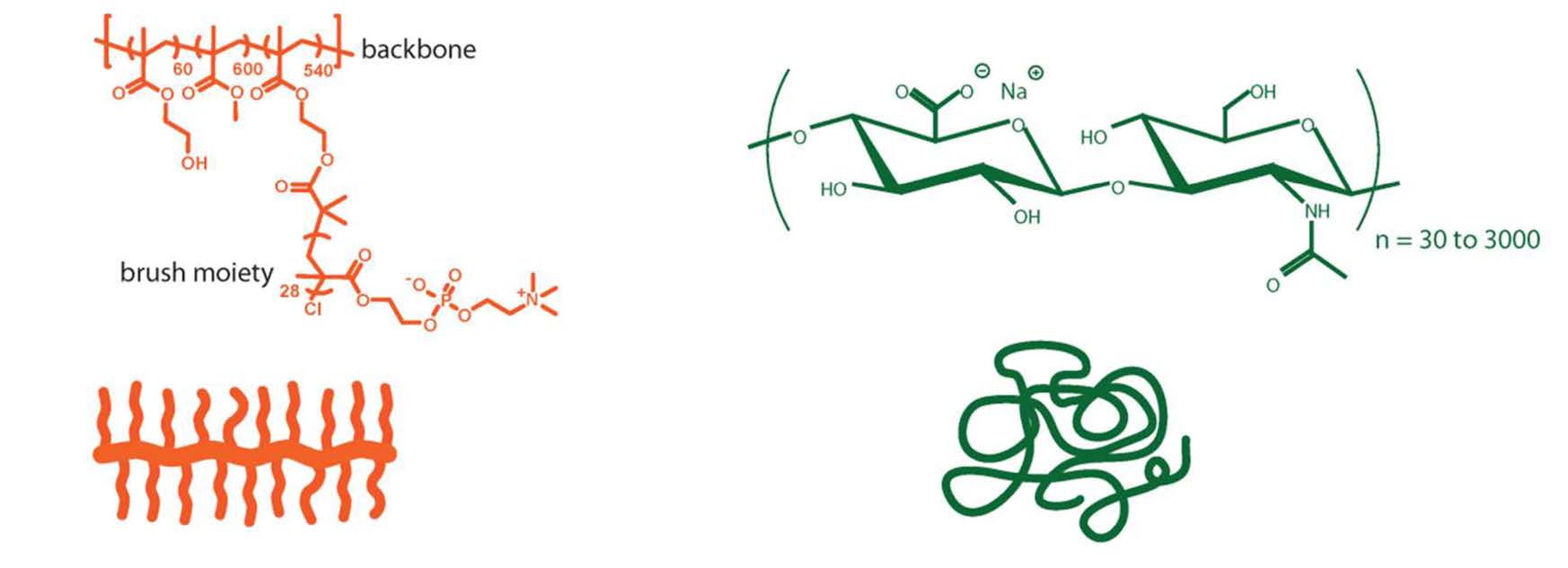
The initial report on development of a bioinspired bottlebrush polymer that exhibits low friction and Amotonons-like behavior was a paper published in 2014 (86) that reported the design of a bottlebrush polymer whose architecture closely mimicked the lubricating protein lubricin, a major component of mammalian synovial fluids. Lubricin can self-associate to form a highly interconnected network. It can also form dimers or multimers whose conformation on a surface can be described as a loop. (87) It is this particular conformation in combination with its bottle-brush architecture that is believed to provide the excellent tribological properties to this protein. A polymer mimic of lubricin was designed based on an ABA triblock polymer, exhibiting a central block with a bottle-brush architecture and two lateral linear blocks designed to provide strong adhesion to the surfaces it adsorbs onto and was expected to promote the formation of brush-polymer loops when adsorbed on surfaces.
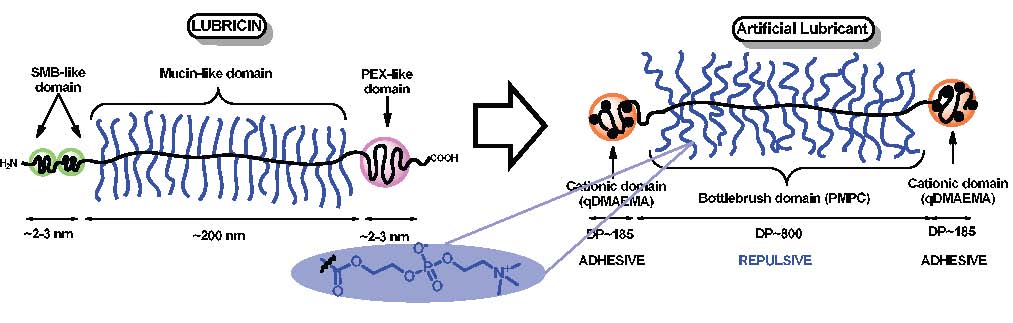
Since the surfaces of study are made of atomically flat mica, the adhesive (A)-blocks of the polymer were composed of a positively charged amine monomer, quaternized 2-(dimethylaminoethyl) methacrylate (qDMAEMA) and a hydrophobic (methyl methacrylate (MMA) monomer. These two components generate polymer segments that strongly adsorb on negatively charged mica surfaces from aqueous solution via electrostatic and hydrophobic interactions. The bottle-brush block of the polymer was composed of a flexible MMA backbone decorated with polyzwitterionic branches of poly(2-methacryloyloxyethyl phosphorylcholine) (PMPC) (shown in blue in the above figure).
Atomic force microscopy (AFM) can resolve the images of individual polymer chains adsorbed on a mica surface from a dilute solution of individual polymer chains. There was a significant difference in organization of the polymer chains when they adsorbed from solutions with different concentrations of the brush- molecules; namely 37 and 370 μg/mL. Rather unexpectedly isolated chains were rarely observed when a concentration of 37 μg/mL was examined, as most of the chains tended to form small aggregates of two to five molecules. While with the higher concentration of 370μg/mL, the surface is almost fully covered by a mono-layer of polymer chains. In the monolayer, the polymer chains tend to maximize possible contacts with their neighboring molecules by adopting a chain extended conformation.

Surface Forces Apparatus (SFA), between two mica surfaces fully covered by the polymer demonstrate that the polymer adopts a loop conformation giving rise to a weak and long-range repulsive interaction force between the surfaces. Under high compression, stronger repulsive forces appear due to the compression of the grafted pendant chains of the polymer. When submitted to shear, the system shows extremely low frictional forces dependent on the salinity of the medium. Friction coefficients measured for this system were as low as ∼10−3. Interestingly, the confined lubricating fluid obeys all three Amontons’ laws. This peculiar observation is a result of the strong shear thinning of the confined fluid and the osmotic repulsive forces that dominate the overall (dynamic and equilibrium) surface interactions.
While the lubrication performance of these ABA block bottlebrush copolymers was exceptional the multi-step synthesis procedure was thought to be a limiting factor to its acceptance by commercial producers. This provided a target of simplification of the synthetic procedure while providing excellent wear protection without any chemical modification of the surfaces. This specific targeted set of properties resulted in development of lubricating and wear protecting fluids based on development of synergistic mixtures of bottlebrushes (BB) and linear polymers solutions. (88)
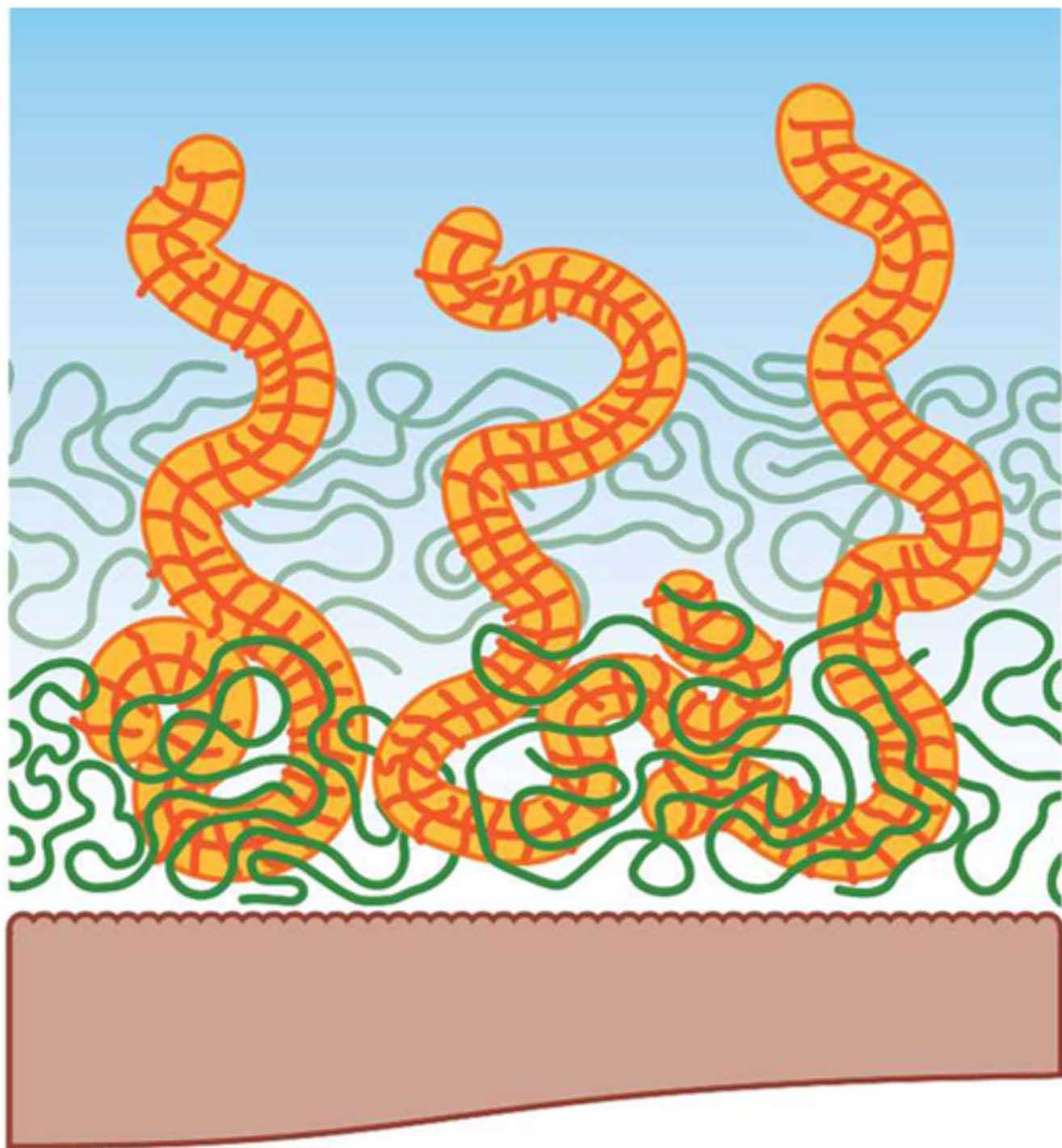
The concept was evaluated by examining the properties of a mixture of hyaluronic acid (HA) - a naturally occurring linear polyelectrolyte, and a water soluble synthetic BB polymer containing similar zwitterionic pendant chains to central block in the ABA block brush copolymer discussed above. Individually, these two polymers exhibit poor wear-protecting capabilities compared to saline solutions but a mixture of the two polymers in pure water, or in saline, provides a dramatic increase in wear protection of surfaces under a wide range of shearing conditions. This synergy between the BB and HA polymers emerges from a strong, yet transient, cohesion between the two components forming the boundary film due to entanglements between both polymers.
A synergistic interaction between a molecular brush macromolecule and a naturally occurring linear polyelectrolyte, hyaluronic acid provides wear protection of surfaces over a wide range of shearing conditions. (88) This synergy between the bottlebrush molecules and the linear hyaluronic acid emerges from a strong cohesive interaction between the two components forming the boundary film due to entanglements between both polymers. Schematics of the brush copolymer and sodium hyaluronic acid used in the study are shown below next to a schematic representation of the formed interfacial polymer layer.
(A) (B) (C)
It was shown that it was possible to design lubricating fluids that were able to provide excellent wear protection without any chemical modification of the surfaces. The fluids use two components, bottlebrush polymers shown in the above scheme as (A) containing zwitterionic pendant chains synthesized by ATRP and a natural linear polymer, sodium hyaluronate scheme as (B). Both components are soluble in pure water or saline conditions. Schematic (C) provides a representation of the formed interfacial polymer layer comprising both polymers.
A surface force apparatus (SFA) was used to characterize the wear protection capacity and the lubricating properties of the fluids. The SFA allows measurement of frictional forces over a wide range of pressure and sliding speeds while monitoring the separation distance between the surfaces at ±0.5 Å resolution as well as the shape of the contact. Muscovite mica was the substrate of choice in SFA experiments mostly because of its optical transparency and atomic flatness and was used due to its extreme propensity to suffer damage under moderate shearing conditions in water and saline conditions, which makes it the perfect substrate to test wear protection. An extensive series of measurements were undertaken and it was determined that the polymer mixtures form an interpenetrated layered thin film represented above in the schematic. The film contains HA molecules adsorbed at the surface and portions of the BB polymer chains extending in the medium. The critical pressure at which the onset of surface damage was triggered was measured and it was found to depend on the molecular weight of the HA when measured in the presence of BB molecules; wear protection properties increased with the molecular weight of the HA increasing from 0 MPa in the absence of BB polymer to 3.2 MPa for HA with a MW of 1500kDA. This observation shows that the cohesive strength of the film depends on chain entanglements for the polymer film cohesion.
This concept can be applied to other types of linear polymers and surfaces and is independent of the chemical and mechanical properties of the surfaces.
The conclusion of the study was that the results indicate that a large range of applications for such fluids can be confidently anticipated. The polymers tested are biocompatible and suitable for intra-articular injection, which suggests a potential use as biolubricants. Other applications requiring wear protection of soft or ductile materials such as plastics (e.g., medical devices) or metals (e.g., articular implants) can also be envisioned.
Furthermore the strategy should also be applicable to non-aqueous systems simply by adapting the chemical functionalities of the BB polymer and the linear polymer counterpart to meet the requirements of the application. On a more fundamental level, the lubricating system described demonstrates that it is possible to control lubrication (i.e., the coefficient of friction μ) and wear protection (P*) independently by tuning the properties of each component.
REFERENCES1. Yu, W. H.; Kang, E. T.; Neoh, K. G.; Zhu, S., Controlled Grafting of Well-Defined Polymers on Hydrogen-Terminated Silicon Substrates by Surface-Initiated Atom Transfer Radical Polymerization. Journal of Physical Chemistry B 2003, 107 (37), 10198-10205.
2. Lee, S. B.; Koepsel, R. R.; Morley, S. W.; Matyjaszewski, K.; Sun, Y.; Russell, A. J., Permanent, Nonleaching Antibacterial Surfaces. 1. Synthesis by Atom Transfer Radical Polymerization. Biomacromolecules 2004, 5 (3), 877-882.
3. Huang, J.; Murata, H.; Koepsel, R. R.; Russell, A. J.; Matyjaszewski, K., Antibacterial Polypropylene via Surface-Initiated Atom Transfer Radical Polymerization. Biomacromolecules 2007, 8 (5), 1396-1399.
4. Huang, J.; Koepsel, R. R.; Murata, H.; Wu, W.; Lee, S. B.; Kowalewski, T.; Russell, A. J.; Matyjaszewski, K., Nonleaching Antibacterial Glass Surfaces via "Grafting Onto": The Effect of the Number of Quaternary Ammonium Groups on Biocidal Activity. Langmuir 2008, 24 (13), 6785-6795.
5. Xing, T.; Hu, W.; Li, S.; Chen, G., Preparation, structure and properties of multi-functional silk via ATRP method. Applied Surface Science 2012, 258 (7), 3208-3213.
6. Xing, T.; Liu, J.; Li, S., Surface-initiated atom transfer radical polymerization on cotton fabric in water aqueous. Textile Research Journal 2013, 83 (4), 363-370, 8 pp.
7. Huang, J.; Cusick, B.; Pietrasik, J.; Wang, L.; Kowalewski, T.; Lin, Q.; Matyjaszewski, K., Synthesis and In Situ Atomic Force Microscopy Characterization of Temperature-Responsive Hydrogels Based on Poly(2-(dimethylamino)ethyl methacrylate) Prepared by Atom Transfer Radical Polymerization. Langmuir 2007, 23 (1), 241-249.
8. Huang, J.; Russell, A. J.; Tsarevsky, N. V.; Matyjaszewski, K. Modification of surfaces with polymers prepared by controlled radical polymerization. WO 2008021500.
9. Dong, H.; Huang, J.; Koepsel, R. R.; Ye, P.; Russell, A. J.; Matyjaszewski, K., Recyclable Antibacterial Magnetic Nanoparticles Grafted with Quaternized Poly(2-(dimethylamino)ethyl methacrylate) Brushes. Biomacromolecules 2011, 12 (4), 1305-1311.
10. Murata, H.; Koepsel, R. R.; Matyjaszewski, K.; Russell, A. J., Permanent, non-leaching antibacterial surfaces-2: How high density cationic surfaces kill bacterial cells. Biomaterials 2007, 28 (32), 4870-4879.
11. Kusumo, A.; Bombalski, L.; Lin, Q.; Matyjaszewski, K.; Schneider, J. W.; Tilton, R. D., High Capacity, Charge-Selective Protein Uptake by Polyelectrolyte Brushes. Langmuir 2007, 23 (8), 4448-4454.
12. Lele, B. S.; Murata, H.; Matyjaszewski, K.; Russell, A. J., Synthesis of Uniform Protein-Polymer Conjugates. Biomacromolecules 2005, 6 (6), 3380-3387.
13. Jones, M.-C.; Ranger, M.; Leroux, J.-C., pH-Sensitive Unimolecular Polymeric Micelles: Synthesis of a Novel Drug Carrier. Bioconjugate Chemistry 2003, 14 (4), 774-781.
14. Nicolas, J.; Mantovani, G.; Haddleton, D. M., Living radical polymerization as a tool for the synthesis of polymer-protein/peptide bioconjugates. Macromol. Rapid Commun. 2007, 28 (10), 1083-1111.
15. Lutz, J.-F.; Boerner, H. G.; Weichenhan, K., 'Click' Bioconjugation of a Well-Defined Synthetic Polymer and a Protein Transduction Domain. Aust. J. Chem. 2007, 60 (6), 410-413.
16. Lutz, J.-F.; Boerner, H. G., Modern trends in polymer bioconjugates design. Prog. Polym. Sci. 2008, 33 (1), 1-39.
17. Oh, J. K.; Drumright, R.; Siegwart, D. J.; Matyjaszewski, K., The development of microgels/nanogels for drug delivery applications. Prog. Polym. Sci. 2008, 33 (4), 448-477.
18. Averick, S.; Mehl, R. A.; Das, S. R.; Matyjaszewski, K., Well-defined biohybrids using reversible-deactivation radical polymerization procedures. Journal of Controlled Release 2015, 205, 45-57.
19. Falatach, R.; Li, S.; Sloane, S.; McGlone, C.; Berberich, J. A.; Page, R. C.; Averick, S.; Konkolewicz, D., Why synthesize protein-polymer conjugates? The stability and activity of chymotrypsin-polymer bioconjugates synthesized by RAFT. Polymer 2015, Ahead of Print.
20. Averick Saadyah, E.; Paredes, E.; Irastorza, A.; Shrivats Arun, R.; Srinivasan, A.; Siegwart Daniel, J.; Magenau Andrew, J.; Cho Hong, Y.; Hsu, E.; Averick Amram, A.; Kim, J.; Liu, S.; Hollinger Jeffrey, O.; Das Subha, R.; Matyjaszewski, K., Preparation of cationic nanogels for nucleic acid delivery. Biomacromolecules 2012, 13 (11), 3445-9.
21. Sumerlin, B. S., Proteins as Initiators of Controlled Radical Polymerization: Grafting-from via ATRP and RAFT. ACS Macro Lett. 2012, 1, 141-145.
22. Nauka, P. C.; Lee, J.; Maynard, H. D., Enhancing the conjugation yield of brush polymer-protein conjugates by increasing the linker length at the polymer end-group. Polym. Chem. 2016, 7 (13), 2352-2357.
23. Oh, J. K.; Siegwart, D. J.; Lee, H.-i.; Sherwood, G.; Peteanu, L.; Hollinger, J. O.; Kataoka, K.; Matyjaszewski, K., Biodegradable Nanogels Prepared by Atom Transfer Radical Polymerization as Potential Drug Delivery Carriers: Synthesis, Biodegradation, in Vitro Release, and Bioconjugation. J. Am. Chem. Soc. 2007, 129 (18), 5939-5945.
24. Heredia, K. L.; Maynard, H. D., Synthesis of protein-polymer conjugates. Organic & Biomolecular Chemistry 2007, 5 (1), 45-53.
25. Kolb, H. C.; Finn, M. G.; Sharpless, K. B., Click chemistry: diverse chemical function from a few good reactions. Angew. Chem., Int. Ed. 2001, 40 (11), 2004-2021.
26. Sumerlin, B. S.; Tsarevsky, N. V.; Louche, G.; Lee, R. Y.; Matyjaszewski, K., Highly Efficient \"Click\" Functionalization of Poly(3-azidopropyl methacrylate) Prepared by ATRP. Macromolecules 2005, 38 (18), 7540-7545.
27. Bontempo, D.; Heredia, K. L.; Fish, B. A.; Maynard, H. D., Cysteine-Reactive Polymers Synthesized by Atom Transfer Radical Polymerization for Conjugation to Proteins. J. Am. Chem. Soc. 2004, 126 (47), 15372-15373.
28. Le Droumaguet, B.; Mantovani, G.; Haddleton, D. M.; Velonia, K., Formation of giant amphiphiles by post-functionalization of hydrophilic protein-polymer conjugates. Journal of Materials Chemistry 2007, 17 (19), 1916-1922.
29. Geng, J.; Mantovani, G.; Tao, L.; Nicolas, J.; Chen, G.; Wallis, R.; Mitchell, D. A.; Johnson, B. R. G.; Evans, S. D.; Haddleton, D. M., Site-Directed Conjugation of \"Clicked\" Glycopolymers To Form Glycoprotein Mimics: Binding to Mammalian Lectin and Induction of Immunological Function. J. Am. Chem. Soc. 2007, 129 (49), 15156-15163.
30. Reyes-Ortega, F.; Parra-Ruiz, F. J.; Averick, S. E.; Rodriguez, G.; Aguilar, M. R.; Matyjaszewski, K.; San Roman, J., Smart heparin-based bioconjugates synthesized by a combination of ATRP and click chemistry. Polym. Chem. 2013, 4 (9), 2800-2814.
31. Golas, P. L.; Matyjaszewski, K., Marrying click chemistry with polymerization: expanding the scope of polymeric materials. Chemical Society Reviews 2010, 39 (4), 1338-1354.
32. Heredia, K. L.; Bontempo, D.; Ly, T.; Byers, J. T.; Halstenberg, S.; Maynard, H. D., In Situ Preparation of Protein-\"Smart\" Polymer Conjugates with Retention of Bioactivity. J. Am. Chem. Soc. 2005, 127 (48), 16955-16960.
33. Broyer, R. M.; Quaker, G. M.; Maynard, H. D., Designed Amino Acid ATRP Initiators for the Synthesis of Biohybrid Materials. J. Am. Chem. Soc. 2008, 130 (3), 1041-1047.
34. Peeler, J. C.; Woodman, B. F.; Averick, S.; Miyake-Stoner, S. J.; Stokes, A. L.; Hess, K. R.; Matyjaszewski, K.; Mehl, R. A., Genetically Encoded Initiator for Polymer Growth from Proteins. J. Am. Chem. Soc. 2010, 132 (39), 13575-13577.
35. Averick, S. E.; Bazewicz, C. G.; Woodman, B. F.; Simakova, A.; Mehl, R. A.; Matyjaszewski, K., Protein-polymer hybrids: Conducting ARGET ATRP from a genetically encoded cleavable ATRP initiator. European Polymer Journal 2013, 49 (10), 2919-2924.
36. Averick, S.; Simakova, A.; Park, S.; Konkolewicz, D.; Magenau, A. J. D.; Mehl, R. A.; Matyjaszewski, K., ATRP under Biologically Relevant Conditions: Grafting from a Protein. ACS Macro Lett. 2012, 1 (1), 6-10.
37. Matyjaszewski, K., Atom Transfer Radical Polymerization (ATRP): Current Status and Future Perspectives. Macromolecules 2012, 45 (10), 4015-4039.
38. Chmielarz, P.; Park, S.; Simakova, A.; Matyjaszewski, K., Electrochemically mediated ATRP of acrylamides in water. Polymer 2015, 60, 302-307.
39. Fodor, C.; Gajewska, B.; Rifaie-Graham, O.; Apebende, E. A.; Pollard, J.; Bruns, N., Laccase-catalyzed controlled radical polymerization of N-vinylimidazole. Polym. Chem. 2016, 7, 6617-6625
40. Cohen-Karni, D.; Kovaliov, M.; Ramelot, T.; Konkolewicz, D.; Graner, S.; Averick, S., Grafting challenging monomers from proteins using aqueous ICAR ATRP under bio-relevant conditions. Polym. Chem. 2017, 8, 3992-3998
41. Fantin, M.; Isse, A. A.; Gennaro, A.; Matyjaszewski, K., Understanding the Fundamentals of Aqueous ATRP and Defining Conditions for Better Control. Macromolecules 2015, 48 (19), 6862-6875.
42. Konkolewicz, D.; Magenau, A. J. D.; Averick, S. E.; Simakova, A.; He, H.; Matyjaszewski, K., ICAR ATRP with ppm Cu Catalyst in Water. Macromolecules 2012, 45 (11), 4461-4468.
43. Simakova, A.; Averick, S. E.; Konkolewicz, D.; Matyjaszewski, K., Aqueous ARGET ATRP. Macromolecules 2012, 45 (16), 6371-6379.
44. Averick, S.; Paredes, E.; Li, W.; Matyjaszewski, K.; Das, S. R., Direct DNA Conjugation to Star Polymers for Controlled Reversible Assemblies. Bioconjugate Chem. 2011, 22 (10), 2030-2037.
45. Averick, S. E.; Dey, S. K.; Grahacharya, D.; Matyjaszewski, K.; Das, S. R., Solid-Phase Incorporation of an ATRP Initiator for Polymer-DNA Biohybrids. Angew. Chem., Int. Ed. 2014, 53 (10), 2739-2744.
46. Matyjaszewski, K.; Jakubowski, W.; Min, K.; Tang, W.; Huang, J.; Braunecker, W. A.; Tsarevsky, N. V., Diminishing catalyst concentration in atom transfer radical polymerization with reducing agents. Proceedings of the National Academy of Sciences 2006, 103 (42), 15309-15314.
47. Averick, S. E.; Paredes, E.; Dey, S. K.; Snyder, K. M.; Tapinos, N.; Matyjaszewski, K.; Das, S. R., Autotransfecting short interfering RNA through facile covalent polymer escorts. J. Am. Chem. Soc. 2013, 135 (34), 12508-12511.
48. Kang, S. M.; Choi, I. S.; Lee, K.-B.; Kim, Y., Bioconjugation of poly(poly(ethylene glycol) methacrylate)-coated iron oxide magnetic nanoparticles for magnetic capture of target proteins. Macromol. Res. 2009, 17 (4), 259-264.
49. Kang, S. M.; Lee, B. S.; Kim, W.-J.; Choi, I. S.; Kil, M.; Jung, H.-j.; Oh, E., Specific binding of streptavidin onto the nonbiofouling titanium/titanium oxide surface through surface-initiated, atom transfer radical polymerization and bioconjugation of biotin. Macromol. Res. 2009, 17 (3), 174-180.
50. Averick, S. E.; Magenau, A. J. D.; Simakova, A.; Woodman, B. F.; Seong, A.; Mehl, R. A.; Matyjaszewski, K., Covalently incorporated protein-nanogels using AGET ATRP in an inverse miniemulsion. Polym. Chem. 2011, 2 (7), 1476-1478.
51. Morille, M.; Passirani, C.; Vonarbourg, A.; Clavreul, A.; Benoit, J.-P., Progress in developing cationic vectors for non-viral systemic gene therapy against cancer. Biomaterials 2008, 29 (24-25), 3477-3496.
52. Cho, H.-Y.; Srinivasan, A.; Hong, J.; Hsu, E.; Liu, S.-G.; Shrivats, A.; Kwak, D.; Bohaty, A. K.; Paik, H.-J.; Hollinger, J. O.; Matyjaszewski, K., Synthesis of Biocompatible PEG-Based Star Polymers with Cationic and Degradable Core for siRNA Delivery. Biomacromolecules 2011, 12 (10), 3478–3486.
53. Allen, T. M.; Cullis, P. R., Science 2004, 303, 1818-1822.
54. Cho, H. Y.; Averick, S. E.; Paredes, E.; Wegner, K.; Averick, A.; Jurga, S.; Das, S. R.; Matyjaszewski, K., Star Polymers with a Cationic Core Prepared by ATRP for Cellular Nucleic Acids Delivery. Biomacromolecules 2013, 14 (5), 1262-1267.
55. Hamilton, A. J.; Baulcombe, D. C., Science 1999, 286, 950-952.
56. Blow, N., Nat. Methods 2009, 6, 305-309.
57. Gao, H.; Matyjaszewski, K., Synthesis of functional polymers with controlled architecture by CRP of monomers in the presence of cross-linkers: From stars to gels. Prog. Polym. Sci. 2009, 34 (4), 317-350.
58. Gao, H.; Matyjaszewski, K., Structural Control in ATRP Synthesis of Star Polymers Using the Arm-First Method. Macromolecules 2006, 39 (9), 3154-3160.
59. Zhang, X.; Xia, J.; Matyjaszewski, K., End-Functional Poly(tert-butyl acrylate) Star Polymers by Controlled Radical Polymerization. Macromolecules 2000, 33 (7), 2340-2345.
60. Cho, H. Y.; Gao, H.; Srinivasan, A.; Hong, J.; Bencherif, S. A.; Siegwart, D. J.; Paik, H.-j.; Hollinger, J. O.; Matyjaszewski, K., Rapid Cellular Internalization of Multifunctional Star Polymers Prepared by Atom Transfer Radical Polymerization. Biomacromolecules 2010, 11 (9), 2199-2203.
61. Park, S.; Cho, H. Y.; Yoon, J. A.; Kwak, Y.; Srinivasan, A.; Hollinger, J. O.; Paik, H.-j.; Matyjaszewski, K., Photo-Cross-Linkable Thermoresponsive Star Polymers Designed for Control of Cell-Surface Interactions. Biomacromolecules 2010, 11 (10), 2647-2652.
62. Oh, J. K.; Tang, C.; Gao, H.; Tsarevsky, N. V.; Matyjaszewski, K., Inverse Miniemulsion ATRP: A New Method for Synthesis and Functionalization of Well-Defined Water-Soluble/Cross-Linked Polymeric Particles. J. Am. Chem. Soc. 2006, 128 (16), 5578-5584.
63. Oh, J. K.; Perineau, F.; Matyjaszewski, K., Preparation of Nanoparticles of Well-Controlled Water-Soluble Homopolymers and Block Copolymers Using an Inverse Miniemulsion ATRP. Macromolecules 2006, 39 (23), 8003-8010.
64. Siegwart, D. J.; Oh, J. K.; Matyjaszewski, K., ATRP in the design of functional materials for biomedical applications. Prog. Polym. Sci. 2012, 37 (1), 18-37.
65. Siegwart, D. J.; Oh, J. K.; Gao, H.; Bencherif, S. A.; Perineau, F.; Bohaty, A. K.; Hollinger, J. O.; Matyjaszewski, K., Biotin-, pyrene-, and GRGDS-functionalized polymers and nanogels via ATRP and end group modification. Macromol. Chem. Phys. 2008, 209 (21), 2179-2193.
66. Oh, J. K.; Siegwart, D. J.; Matyjaszewski, K., Synthesis and Biodegradation of Nanogels as Delivery Carriers for Carbohydrate Drugs. Biomacromolecules 2007, 8 (11), 3326-3331.
67. Siegwart, D. J.; Bencherif, S. A.; Srinivasan, A.; Hollinger, J. O.; Matyjaszewski, K., Synthesis, characterization, and in vitro cell culture viability of degradable poly(N-isopropylacrylamide-co-5,6-benzo-2-methylene-1,3-dioxepane)-based polymers and crosslinked gels. J. Biomed. Mater. Res., Part A 2008, 87A (2), 345-358.
68. Bencherif Sidi, A.; Srinivasan, A.; Horkay, F.; Hollinger Jeffrey, O.; Matyjaszewski, K.; Washburn Newell, R., Influence of the degree of methacrylation on hyaluronic acid hydrogels properties. Biomaterials 2008, 29 (12), 1739-49.
69. Bencherif, S. A.; Siegwart, D. J.; Srinivasan, A.; Horkay, F.; Hollinger, J. O.; Washburn, N. R.; Matyjaszewski, K., Nanostructured hybrid hydrogels prepared by a combination of atom transfer radical polymerization and free radical polymerization. Biomaterials 2009, 30 (29), 5270-5278.
70. Siegwart, D. J.; Srinivasan, A.; Bencherif, S. A.; Karunanidhi, A.; Oh, J. K.; Vaidya, S.; Jin, R.; Hollinger, J. O.; Matyjaszewski, K., Cellular Uptake of Functional Nanogels Prepared by Inverse Miniemulsion ATRP with Encapsulated Proteins, Carbohydrates, and Gold Nanoparticles. Biomacromolecules 2009, 10 (8), 2300-2309.
71. Oh, J. K.; Bencherif, S. A.; Matyjaszewski, K., Atom transfer radical polymerization in inverse miniemulsion: A versatile route toward preparation and functionalization of microgels/nanogels for targeted drug delivery applications. Polymer 2009, 50 (19), 4407-4423.
72. Richard, R. E.; Schwarz, M.; Ranade, S.; Chan, A. K.; Matyjaszewski, K.; Sumerlin, B., Evaluation of Acrylate-Based Block Copolymers Prepared by Atom Transfer Radical Polymerization as Matrices for Paclitaxel Delivery from Coronary Stents. Biomacromolecules 2005, 6 (6), 3410-3418.
73. Richard, R. E.; Schwarz, M.; Ranade, S.; Chan, A. K.; Matyjaszewski, K.; Sumerlin, B., Acrylate-based block copolymers prepared by atom transfer radical polymerization as matrices for drug delivery applications. ACS Symp. Ser. 2006, 944 (Controlled/Living Radical Polymerization), 234-251.
74. Cordeiro, R. A.; Rocha, N.; Mendes, J. P.; Matyjaszewski, K.; Guliashvili, T.; Serra, A. C.; Coelho, J. F. J., Synthesis of well-defined poly(2-(dimethylamino)ethyl methacrylate) under mild conditions and its co-polymers with cholesterol and PEG using Fe(0)/Cu(ii) based SARA ATRP. Polym. Chem. 2013, 4 (10), 3088-3097.
75. Tsarevsky, N. V.; Matyjaszewski, K., Reversible Redox Cleavage/Coupling of Polystyrene with Disulfide or Thiol Groups Prepared by Atom Transfer Radical Polymerization. Macromolecules 2002, 35 (24), 9009-9014.
76. Gao, H.; Tsarevsky, N. V.; Matyjaszewski, K., Synthesis of Degradable Miktoarm Star Copolymers via Atom Transfer Radical Polymerization. Macromolecules 2005, 38 (14), 5995-6004.
77. Tsarevsky, N. V.; Matyjaszewski, K., Combining Atom Transfer Radical Polymerization and Disulfide/Thiol Redox Chemistry: A Route to Well-Defined (Bio)degradable Polymeric Materials. Macromolecules 2005, 38 (8), 3087-3092.
78. Brown, J. M.; Wilson, W. R., Exploiting tumor hypoxia in cancer treatment. Nat. Rev. Cancer 2004, 4 (6), 437-447.
79. Lutz, J.-F.; Boerner, H. G.; Weichenhan, K., Combining Atom Transfer Radical Polymerization and click chemistry: A versatile method for the preparation of end-functional polymers. Macromolecular rapid communications 2005, 26 (7), 514-518.
80. Averick, S. E.; Paredes, E.; Irastorza, A.; Shrivats, A. R.; Srinivasan, A.; Siegwart, D. J.; Magenau, A. J.; Cho, H. Y.; Hsu, E.; Averick, A. A.; Kim, J.; Liu, S.; Hollinger, J. O.; Das, S. R.; Matyjaszewski, K., Preparation of Cationic Nanogels for Nucleic Acid Delivery. Biomacromolecules 2012, 13 (11), 3445-3449.
81. Mantha Venkat, R. R.; Nair Harsha, K.; Venkataramanan, R.; Gao Yuan, Y.; Matyjaszewski, K.; Dong, H.; Li, W.; Landsittel, D.; Cohen, E.; Lariviere William, R., Nanoanesthesia: A Novel, Intravenous Approach to Ankle Block in the Rat by Magnet-Directed Concentration of Ropivacaine-Associated Nanoparticles. Anesthesia and analgesia 2014.
82. Dong, H.; Mantha, V.; Matyjaszewski, K., Thermally Responsive PM(EO)2MA Magnetic Microgels via Activators Generated by Electron Transfer Atom Transfer Radical Polymerization in Miniemulsion. Chem. Mater. 2009, 21, 3965-3972.
83. Pelegri-O'Day, E. M.; Lin, E.-W.; Maynard, H. D., Therapeutic Protein-Polymer Conjugates: Advancing Beyond PEGylation. J. Am. Chem. Soc. 2014, 136 (41), 14323-14332.
84. Prausnitz, M. R.; Langer, R. Nat. Biotechnol. 2008, 26, 1261−1268.
85. Li, S.; Chung Hee, S.; Simakova, A.; Wang, Z.; Park, S.; Fu, L.; Matyjaszewski, K.; Cohen-Karni, D.; Averick, S., Biocompatible Polymeric Analogues of DMSO Prepared by Atom Transfer Radical Polymerization. Biomacromolecules 2017, 18 (2), 475-482.
86. Banquy, X.; Burdynska, J.; Lee, D. W.; Matyjaszewski, K.; Israelachvili, J., Bioinspired Bottle-Brush Polymer Exhibits Low Friction and Amontons-like Behavior. J. Am. Chem. Soc. 2014, 136 (17), 6199-6202.
87. Zappone, B.; Greene George, W.; Oroudjev, E.; Jay Gregory, D.; Israelachvili Jacob, N., Molecular aspects of boundary lubrication by human lubricin: effect of disulfide bonds and enzymatic digestion. Langmuir 2008, 24 (4), 1495-508.
88. Faivre, J.; Shrestha, B. R.; Burdynska, J.; Xie, G.; Moldovan, F.; Delair, T.; Benayoun, S.; David, L.; Matyjaszewski, K.; Banquy, X., Wear Protection without Surface Modification Using a Synergistic Mixture of Molecular Brushes and Linear Polymers. ACS Nano 2017, 11 (2), 1762–1769.