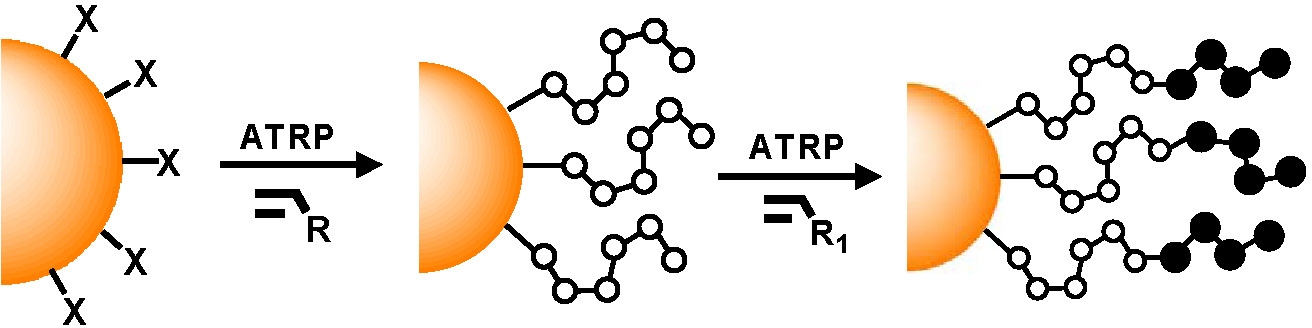
Spherical Particles
Several procedures have been developed for the preparation of hybrid materials based on a suitably functionalized inorganic core material. They include the most common procedure discussed in detail below, “grafting from” (1, 2, 3) but also “grafting onto” and "grafting through" (4, 5) ‘templated synthesis (6, 7) and “ligand exchange” (8) have also been successfully employed form preparation of well-defined hybrid particles. A recent review, discussing all procedures including grafting through and all surface geometries, is provided in reference. (9, 10)
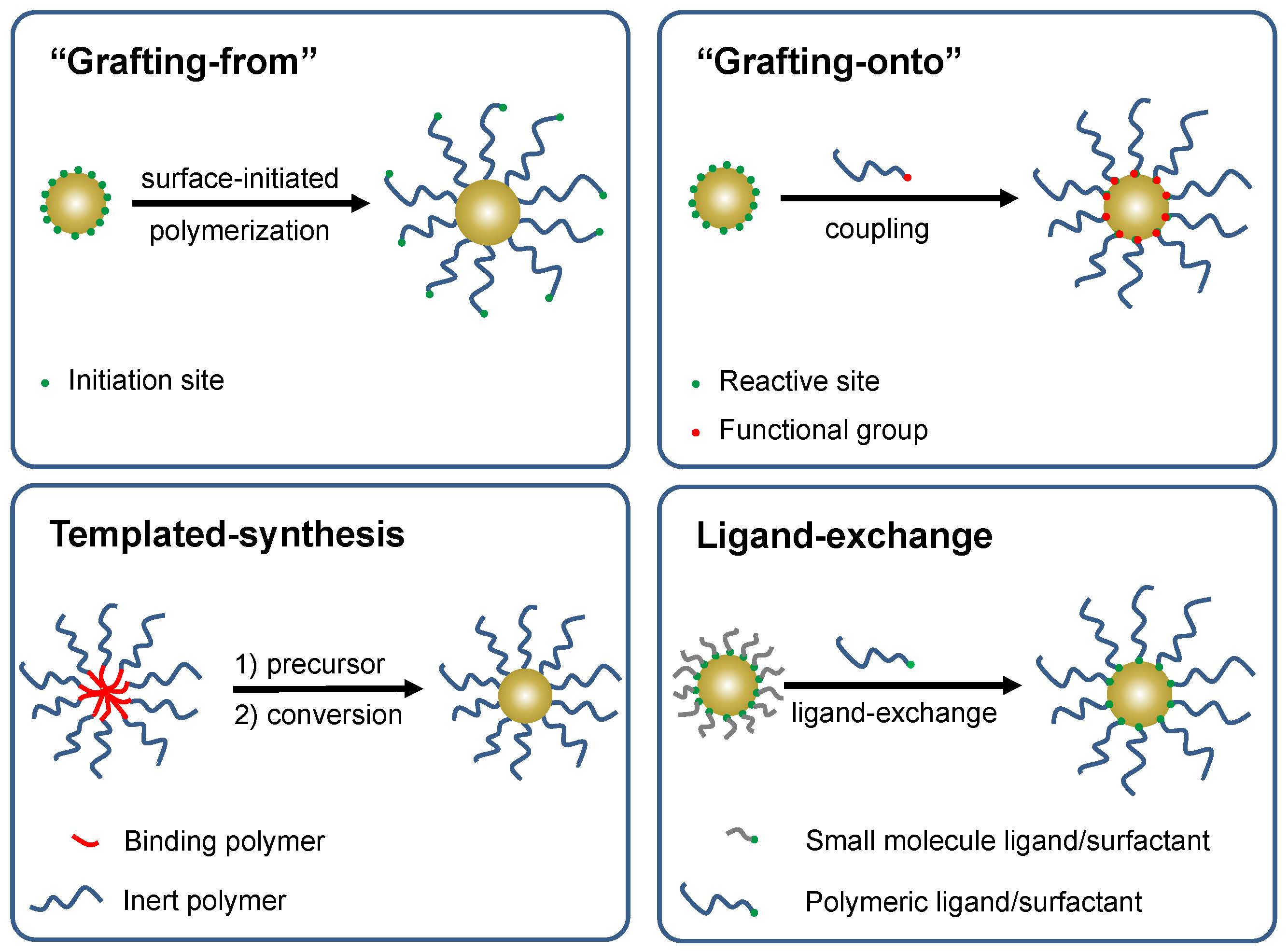
The functionalization, or utilization of the functional groups inherently present on the surfaces, of many solids; including silica (SiO2), gold, silver, germanium, lead sulfide, carbon black, iron oxides and many other metal oxide systems has been achieved, (3, 11, 12) allowing attachment of initiators for an ATRP of many monomers forming organic/inorganic hybrid nanoparticles containing an inorganic core and a tethered functional polymeric shell selected for possible application in nano-electronics, nano-photonics, catalysis, nano-patterning, drug delivery, and bio-sensing. (12)
Procedures for surface functionalization
Earlier papers and review articles (2, 11, 13, 14) detail procedures for the preparation of well-defined polymer-nanoparticle hybrid materials by modifying the surface of particles of different composition and different sizes with an ATRP initiator via different surface chemistries depending on the type of targeted surface. In the case of gold and silica particles by tethering functional thiols or chloro/alkoxy silanes respectively, e.g. in one example silica particles were functionalized with (2-(4-chloromethylphenyl)ethyl) dimethylethoxysilane. (15)
Recent papers have examined procedures for enhancing initiation efficiency. One paper described utilizing a photoinduced metal free procedure (16) and the other an examination of the influence of the length of the spacer between the anchoring group and initiating functionality in SI-ATRP. (17) The motivation for the first study was the limited initiation efficiency when 2-bromoisobutyrate (BiB) was used as the initiator (18) and this was overcome by selection of ethyl 2-bromo-2-phenylacetate (EBPA) as a more reactive initiator. The initiating functionality was linked to two different silane anchoring groups to create novel tetherable initiators, which were then compared to conventional BiB-based tetherable initiators. Silica nanoparticles with diameters of 16 nm and 120 nm were used as the inorganic core for the SI-ATRP reactions. Chlorodimethylsilane was used as anchoring group for the 16 nm silica nanoparticles to avoid formation of multiple layers and a triethoxysilane moiety was used for the 120 nm silica nanoparticles to ensure more stable bonding. Six metal-free SI-ATRP reactions were performed to compare the influence of different parameters, such as initiator type, particle size, and catalyst concentration on the “grafting from” polymerizations. The accessible initiation sites for surface-initiated polymerization depend upon various reaction parameters, including bulkiness of monomers and initiation efficiency. (19) A high target degree of polymerization (DP) of 500 was chosen in order to provide a dilute reaction medium and avoid interparticle cross-linking. (3) Polymerization from 16 nm silica particles functionalized with 6-(chlorodimethylsilyl)hexyl 2-bromo-2-phenylacetate (BPASiCl) displayed a progressive increase in molecular weight (MW) and approached the theoretical values as well as generating grafted polymers with a low Ð. The previous report on metal-free SI-ATRP used a significantly higher catalyst concentration (10 mg/mL) and achieved a moderate Ð for the polymers obtained with BiB-based tetherable initiator. (18) This indicated that a higher catalyst concentration might mitigate the slow initiation and improve graft density. Therefore, two experiments, with higher concentrations of catalyst (1.3 mg/mL), were performed to evaluate the influence of catalyst concentration. Both reactions with higher catalyst concentrations displayed a significant improvement in control over the polymerization and higher graft densities. A linear semilogarithmic kinetic plot indicated efficient initiation and both the regular increase of MW and a decrease of Ð with conversion, demonstrating good control over the polymerization and providing a final product with graft density of 0.60 nm−2. TEM images confirmed successful grafting of PMMA from silica nanoparticles and showed a small fraction of aggregated nanoparticles. In conclusion the BPA-based tetherable initiators are capable of initiating metal-free SI-ATRP, and should be also applicable to SI-ATRP with Cu-catalyzed systems.
TEM images of PMMA grafted 16 nm silica particles showed only a small fraction of aggregated nanoparticles formed with a ratio of reagents: [SiO2−Br, ∼1 Br/nm2]0/[MMA]0/[PhPTZ]0 = 1:500:0.1/0.5; DMA = 50 vol %; under UV irradiation at 365 nm at room temperature over 3.0 h reaction reaching 36% conversion and forming grafted chains with Mn = 4.70 x 104 and Ð = 1.62.
In the paper detailing the influence of spacers in the tetherable initiators on SI-ATRP (17) tetherable initiators bearing a chlorodimethylsilyl anchoring group and a 2-bromoisobutyrate initiating group with three different alkyl spacers, C3, C6, and C11, were synthesized using three commercially available vinyl alcohols as starting materials.
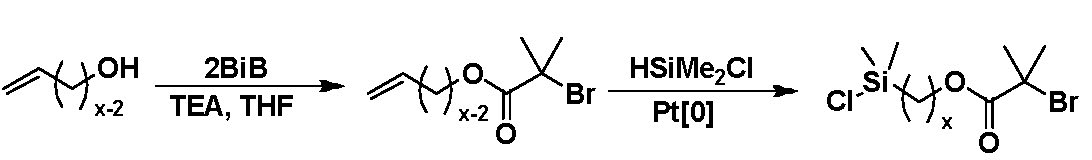
SiO2 NPs (ca. 16 nm diameter) were surface-modified with the three tetherable initiators, resulting in three batches of functionalized SiO2 NPs with apparent bromine densities of ∼1.5 Br/nm2.
Normal, photo- and metal free ATRP were employed in the grafting from polymerizations for the three tethered initiators. To improve the control of polymerization, halogen exchange was used in normal ATRP to mitigate the influence of penultimate unit effect (20) and excess ligand, Me6TREN was added to act as, and serve as, the sacrificial activator regenerator in photoATRP with 100 ppm of added Cu(II) catalyst, (21) whereas the metal-free ATRP was controlled by a photoredox reaction of 10-phenylphenothiazine (PhPTZ). (22)
Polymerization kinetics and graft densities were measured using the following ratios of reagents:
- [SiO2−Br, ∼1 Br/nm2]0/[MMA]0/[CuCl]0/[CuCl2]0/[dNbpy]0 = 1/500/0.475/0.025/1; 50 vol % anisole; 60 °C for normal ATRP;
- [SiO2−Br]0/[MMA]0/[CuBr2]0/[Me6TREN]0 = 1/500/0.05/0.3; 50 vol % anisole; room temperature, UV irradiation at 365 nm for photo ATRP; and
- [SiO2−Br]0/[MMA]0/[PhPTZ]0 = 1/500/0.5; 50 vol % DMA; room temperature, UV irradiation at 365 nm for metal-free ATRP.
The polymerizations were conducted until the 25 mm × 12 mm oval rare earth extra power stir bar stopped moving. Beyond this point, homogeneity of the reactions could not be ensured, and irreversible interparticle cross-linking might start. An influence of the spacer lengths on kinetics and outcomes of SI-ATRP were demonstrated. In both normal ATRP and metal-free-ATRP, the presence of a longer spacer resulted in more accessible alkyl halide and hence higher graft density additionally forming grafted polymers with narrow dispersity, Ð = ~1.18.
A broadly applicable procedure for tethering ATRP initiators to metal oxide surfaces involves use of 12-(2-bromoisobutyramido)dodecanoic acid (BiBADA), based on fatty acid derivative ω-aminolauric acid, a monomer used at the industrial scale for the preparation of nylon 12. BiBADA was used as a tetherable initiator for a large range of metal oxide nanoparticles including Co3O4, NiO, Y2O3, ZrO2, La2O3, CeO2, WO3, In2O3, Sb2O3 and α-Al2O3. (23)
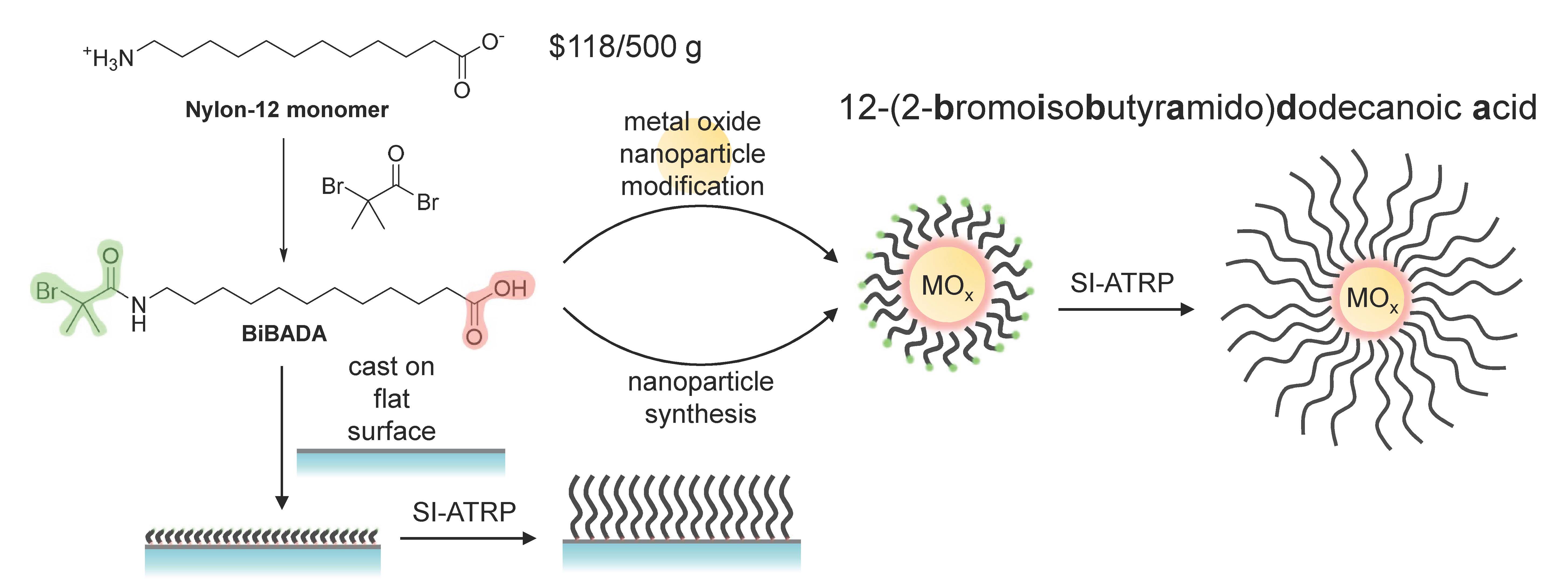
The utility of this method for functionalization of particles was exemplified by the preparation of superparamagnetic iron oxide nanoparticles (SPION) with poly[oligo(ethyleneglycol)methyl ether acrylate-480] (POEGA). The grafted particles were readily dispersible in water and their magnetic relaxivities were measured and compared to FerahemeTM the only FDA approved iron oxide nanoparticles approved for human use. The relaxivity ratios were ten times higher than the reference sample and are therefore highly efficient contract agents for T2* weighted MRI, (23) and were confirmed to be non-toxic, hence effective MRI contrast agents.
The “universal” tetherable initiator was also employed for funtionalization of a range of flat surfaces inclding gibbsite nanoplatelet particles allowing grafting from PMMA brushes with grafting densities higher than 0.4 nm-2 with minimum amount of free polymer and narrow Ð. (24) Such nanopaticles provide oppertunities for enhancement of mechanical, barrier and optical properties of composite materials arising from the shape anisopropic disc like morphology of the distributed particle fillers. Using BIBADA as initiator for SI-ATRP facilitated formation of gibsonite-g-PMMA particles with high grafting density,-0.4 nm-2, and 12% inorganic content that self organize into a ono-component hybrid material with nacre-like morphology, formd by stacking of nanoplatelet brushes, and mechanical properties the rival high MW PMMA. The glass transition of the tethered PMMA was 126.3 oC which was attributed to increased steric hinderance in the platelet brushes.
Procedures to limit effect of termination reactions
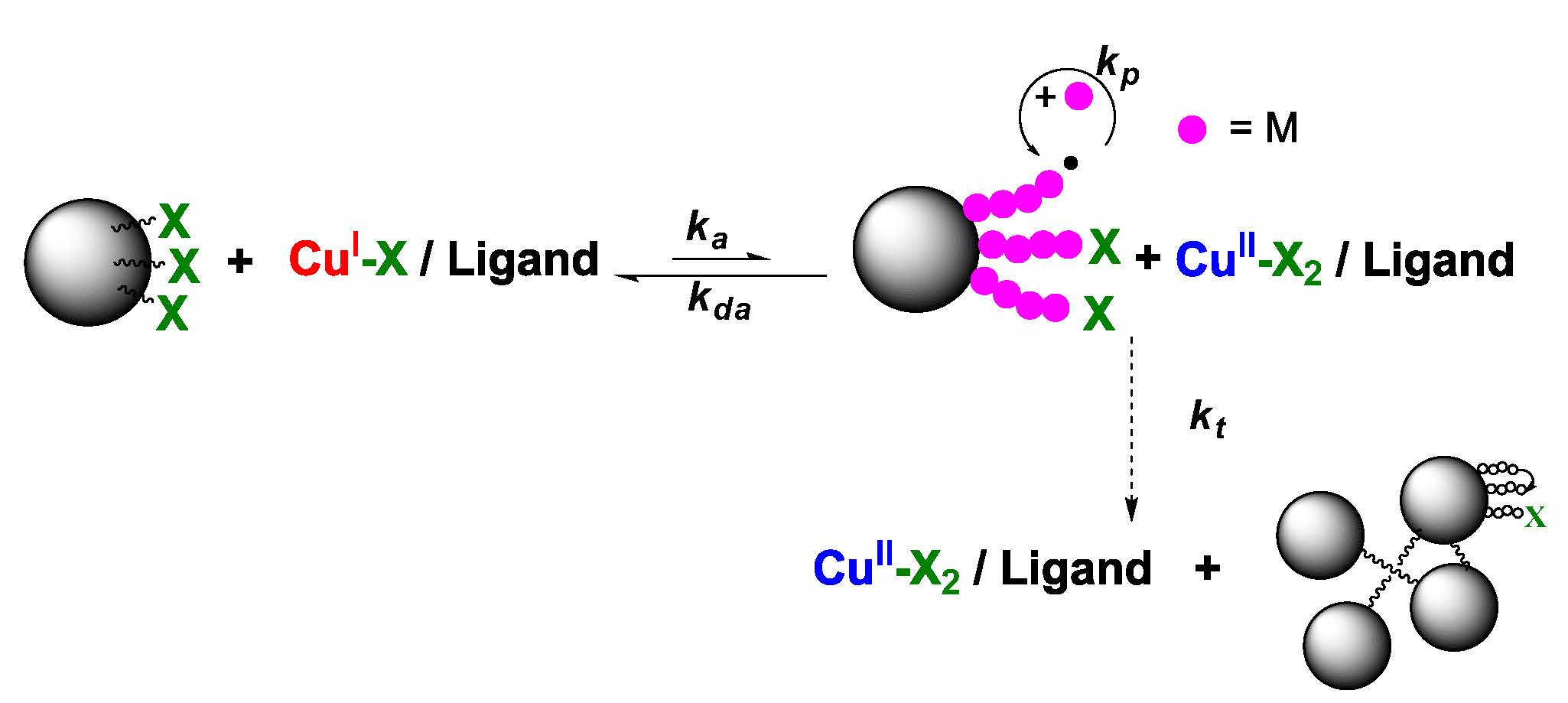
The consequence of radical-radical termination is more serious during the preparation of colloidal particles than in a normal ATRP. In traditional ATRP reactions termination leads to coupling whereas with a particle with 1000’s of initiation sites this leads to interpartile crosslinking; as a result of:
Pc= 1/ {r(fa -1)(fb -1)}½ → 2/f → 1/1000.
So gelation is predicted at 0.1% intermolecular coupling.
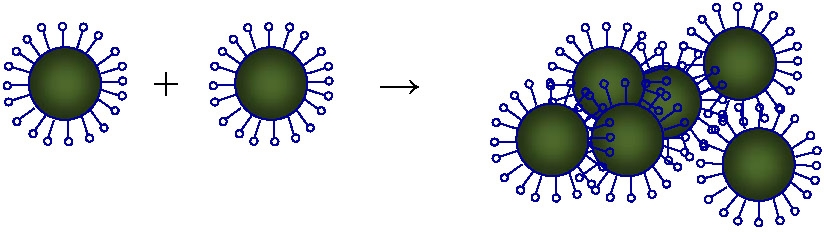
Gelation can be avoided by running the reaction under high dilution conditions to low conversion, i.e. under conditions where there is a low concentration of active radicals and consequently slow propagation. (25) During surface-initiated atom transfer radical polymerization (SI-ATRP) of styrene from the silica nanoparticles, thermal self-initiation of styrene produces unattached polymer chains. Size exclusion chromatography afforded a facile approach to quantify the mass of the unattached polymer, and provided an incentive to develop a substantial refinement to estimates of chain graft density beyond traditionally used approaches, such as thermosgravimetry. This fraction of unattached polymer is still present in the solid polymer/hybrid even after post-polymerization work-up via precipitation and re-dissolution. Removal necessitates additional procedures, such as high speed centrifugation. Selection of a lower polymerization temperature, in concert with a more reactive Cu complex, significantly reduces the amount of unattached polystyrene impurity. (26) Size exclusion chromatography was capable of detecting and quantifying the fraction of unattached polystyrene in the presence of the hybrid particle. Under some circumstances the overestimate of grafting density coiuld be as high as 100% and therefor influenced the prediction and understanding of the properties of the hybrid particle. Changing from running the reaction at 90 0C with a diNbpy ligand to 70 0C and PMDETA reduced the % unattached polystyrene from 17.2% to 4.2% with the same degree of initiation efficiency and a decrease in reaction time from 47 to 33 hours. The improved polymerization conditions and post-polymerization purification provide more refined polystyrene-grafted silica nanoparticles to clarify structure-property relationships of these core/shell hybrids.
Another approach, specifically tailored for the synthesis of hybrid particles with higher molecular weight grafted chains, is synthesis carried out under high pressure. (27)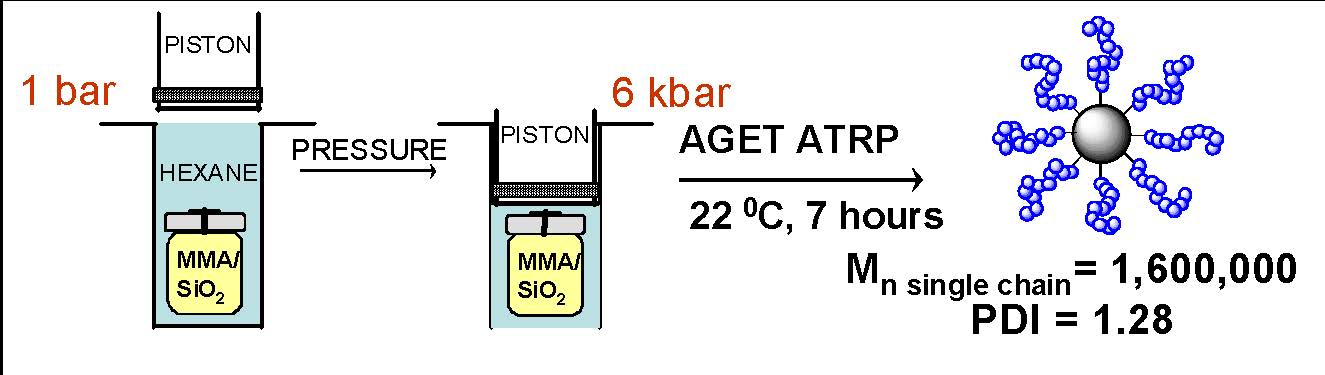
AGET ATRP was used for the polymerization and it was possible to increase the overall rate of polymerization due to the fact that under high pressure the rate of propagation is enhanced while the rate of termination is decreased compared to ATRP at ambient pressure. Two different sized particles were used for the reaction, one 20 nm in diameter and the other 120 nm and two different initiators were tethered to the surface by reacting respectively 1-chlorodimethylsilyl)propyl 2-bromoisobutyrate or 2-bromo-isobutyryloxyhexyltriethoxysilane with the silanol groups on the silica particles surface. On the basis of elemental analysis, 20 nm functional silica particle had average ~2600 initiating sites (~ 2 Br/nm2), whereas 120 nm particle had 105 sites (also ~ 2 Br/nm2). Lower initiator density (~600 initiating sites (0.5 Br/nm2) per 20 nm silica particle) was obtained when chlorotrimethylsilane was used in a mixture with 1-(chlorodimethylsilyl)propyl 2-bromoisobutyrate to block some silanol groups and reduce initiator density. Several polymerizations were carried out at 6 kbars pressure using the following reaction conditions: [MMA]0/[-Br]0/[CuBr2]0/[TPMA]0/[AsAc]0 = 10,000/1/2/25/25 in anisole (46 vol%) and DMF (8 vol%) at 22 °C, where TPMA is tris(2-pyridylmethyl)amine (ligand), AsAc is ascorbic acid (reducing agent) and DMF is dimethylformamide. After the reaction was completed the polymers were detached from silica surface by dissolving silica with HF, and analyzed by SEC. The entire molecular weight curve moved smoothly towards higher MW, with no observable tailing, demonstrating good control over polymerization. The grafting densities of polymer chains were calculated based on the known Mnth and the amount of polymer from thermogravimetric analysis. The highest grafting density of 0.3 chain/nm2, for the brushes with Mn = 1,600,000, suggests that brushes can be considered as “semi-diluted density” brushes. (28)
Miniemulsion: The first example of the successful synthesis of hybrid nanoparticles using multifunctional silica initiators in a miniemulsion ATRP was disclosed a decade ago. The reaction was driven to higher conversion in a shorter time. (29) This success with miniemulsion relies on the compartmentalization of the reaction media which segregates the reaction medium and therefore minimizes the effect of radical termination and macroscopic gelation. (30) Since termination reactions should be limited to individual droplets the proportion of terminated chains should be relatively small and the degree of crosslinking is less than in bulk conditions. The confinement effect increases the deactivation rate and termination rate so control is better but reaction rate is also decreased.
As shown in the AFM image below each silica particle is evenly coated with polymer and the individual particles are not crosslinked even though conversion was driven to greater than 60%. The particles were prepared with a 200:1 monomer to initiator ratio and each n-BA tethered chain has an average Mn=15,900 g/mol.

SiO2-g-pSt hybrid nanoparticles with each tethered polystyrene possessing molar masses in the range of Mn = 5,000 to 33,000 g/mol were prepared using commercially available silica nanoparticles as colloidal initiators, which greatly facilitated scale-up synthesis. The hybrid particles were characterized both in the solid state and in solution using transmission electron microscopy (TEM) and dynamic light scattering (DLS) respectively. TEM images of the SiO2-g-pSt colloids revealed the formation of (sub)monolayer patches with interparticle spacing that increased with an increase in the molar mass of the tethered polystyrene. Comparison of the hydrodynamic radii (Rh) of hybrid nanoparticles of varying size determined by DLS in toluene, versus the molar mass (Mn) of the polystyrene chains cleaved from colloids, determined by SEC, revealed a linear relationship. Such a linear dependence of Rh vs. Mn is a strong indication that when the particles are dispersed in toluene, the tethered chains adopt highly chain extended conformations, presumably due to steric interactions caused by the high grafting density. (31)
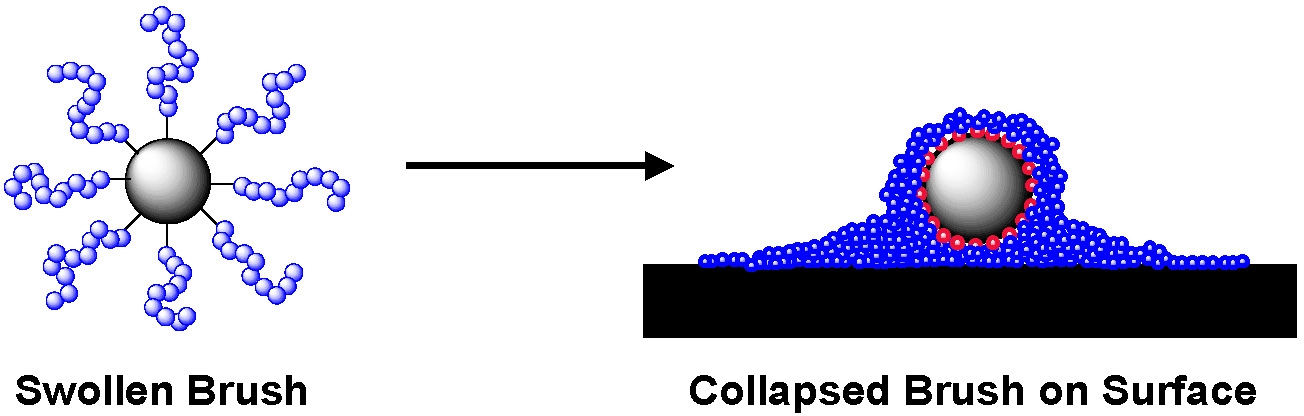
AFM examination of a relatively low concentration of spherical brushes formed by grafting n-butyl acrylate from silica particles deposited on mica surfaces show that the swollen brush has collapsed and that the hard silica core is surrounded by the soft grafted chains.
Since the polymerization from the particle surface was being conducted by ATRP, it is simple task to isolate the particles and add them to a fresh monomer solution and form tethered block copolymers.
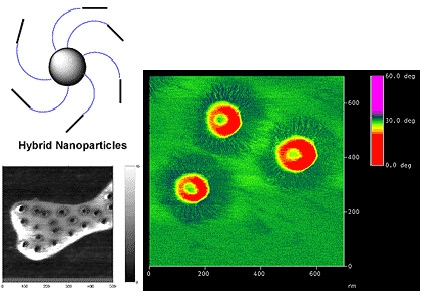
Tri-phasic separation was clearly observed by AFM examination of a particle with tethered poly(St-b-nBA) block copolymer chains. A hard polystyrene core surrounding the silica particle and the soft acrylate shell strongly adsorbed on the mica substrate are observed forming three distinct observable phase separations.
As noted above, conducting the “grafting from” reaction in miniemulsion systems provide some additional benefits. The reactions follow first order kinetics and can be driven at a higher rate to higher conversion without excessive production of coupled particles. Equivalent polymerization rates were observed for miniemulsion reactions initiated by alkyl halides, regardless of whether the initiators were attached to particle surfaces or free in solution. Therefore, the procedure provides a viable commercial approach to novel, functionally-designable materials with properties that can be pre-selected to target many specific applications.[32]
This approach is currently being applied to other multifunctional initiators, including multi-arm star molecules, molecular brushes, and other well-defined polymers with complex architectures. Indeed molecular brushes were successfully synthesized in a miniemulsion system via AGET ATRP.[34] Macroscopic gelation was observed for bulk ATRP but was not detected in miniemulsion. The side-chain polymers grew from backbones rapidly in miniemulsion droplets and high monomer conversion was reached in relatively short time. Molecular visualization by AFM proved that some cross-linking did occur in miniemulsion droplets when the conversion was high (84%). However, this cross-linking showed no effect to the miniemulsion stability and fluidity, and therefore, the synthesized materials can be easily processed for further uses/applications.
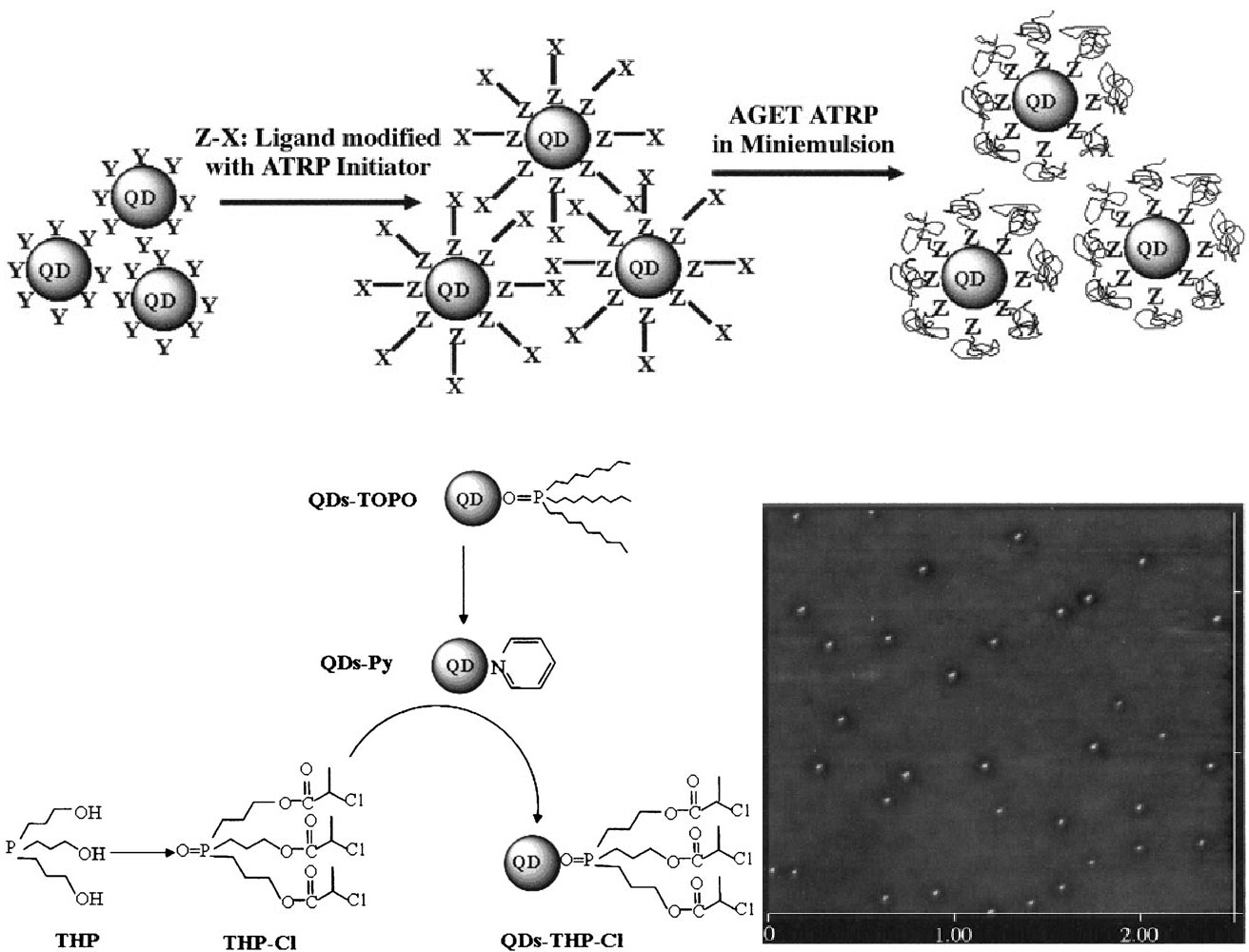
The miniemulsion procedure has also been extended to functionalizing CdS quantum dots with poly(n-butyl acrylate) using activators generated by electron transfer (AGET) ATRP.[30] The quantum dots were first modified by complexation with a phosphorous-containing ligand, which was then further modified to contain an ATRP initiating group. Bromine-containing functionalities degraded the QDs, but a chlorine-functionalized initiating group slowed the degradation enough that 3-4 nm QDs were obtained post-modification. The polymerization of n-butyl acrylate further protected the QDs from degradation via chemical reaction, and the resulting materials were both well-distributed on the nanoscale and possessed the optical properties expected from quantum dots of their size.
A one-pot synthesis of thermally stable core/shell gold nanoparticles (Au-NPs) was developed via surface-initiated atom transfer radical polymn. (ATRP) of Bu acrylate (BA) and a dimethacrylate-based cross-linker.[31]
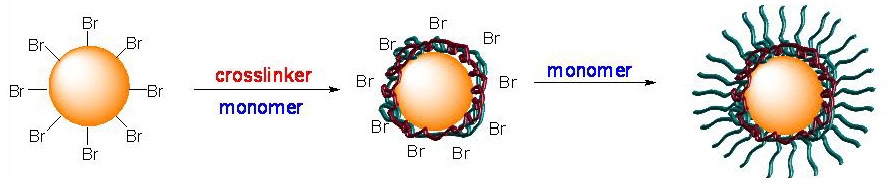
The higher reactivity of the cross-linker enabled the formation of a thin cross-linked polymer shell around the surface of the Au-NP before the growth of linear polymer chains from the shell. The cross-linked polymer shell served as a robust protective layer that prevented the dissociation of linear polymer brushes from the surfaces of Au-NPs, and provided excellent thermal stability to the Au-NPs at elevated temp. (e.g., 110 DegC for 24 h).
The grafting from initiator functionalized particles method could be easily expanded for preparation of other types of inorganic/polymer nanocomposites with significantly improved stability. Indeed the opportunities for designing novel functional materials derive from the vast parameter space that is available for tailoring the interactions between particle brushes, their self-assembly, and hence the properties of particle brush-based materials. Despite their complexity, a common feature of particle brush materials is that properties are intimately related to the conformation of the tethered chains. The latter can be (approximately) described on the basis of mean field models that analyze the implication of chain crowding on the structure and interactions of tethered chains. A detailed discussion of the basis of the transition of the grafted chains from a concentrated particle brush (CPB) regime to a semi-dilute particle brush (SDPB) regime is provided in references (3, 26, 34, 35) and show how the structure and interaction in particle brush materials sensitively depends on the competing influence of stretched and relaxed segments, the DC model provides valuable information for the design of particle brush materials with tailored properties. In a single component system, where the brush forms the matrix, the grafting density + MW + particle sizes influence the brush architecture and hence matrix forming interactions between adjacent polymer brushes. Brushes within the CPB provide higher Tg, but the mechanical properties are not enough to generate utility in any applications. On the other hand once the brushes enter the SDPB the bulk hybrid particle system shows fracture toughness 7 times higher than athermal systems at similar inorganic loadings. This performance enhancement can be attributed to interparticle brush interactions providing flexibility and toughness.
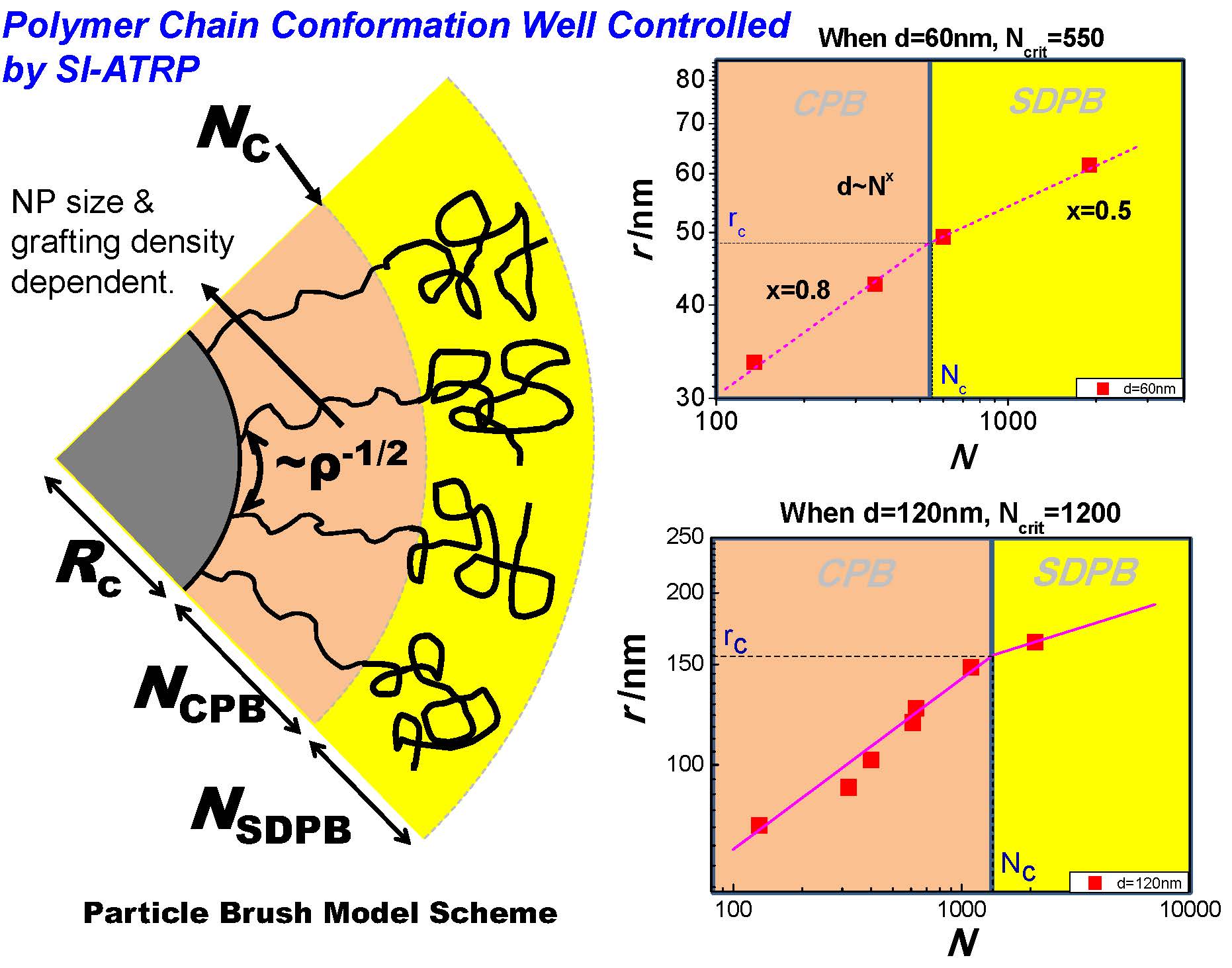
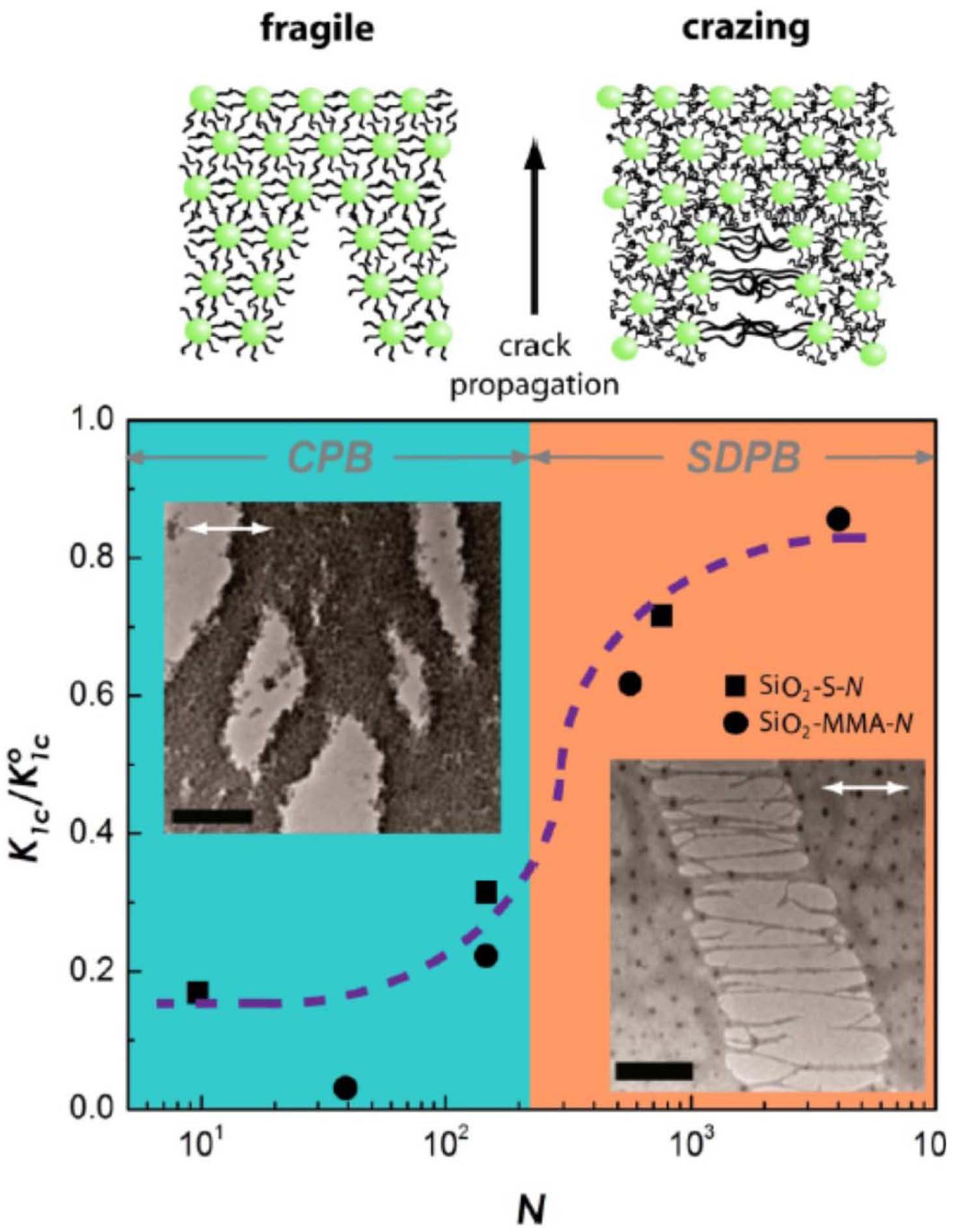
The lower schematic shows the organizational transition that results in a transformation from a fragile matrix material to one that exhibits crazing and hence an enhanced capacity for energy absorption when under stress.
Dependence of reduced fracture toughness Kic/Kic0 on the degree of polymerization N of grafted chains for PS-grafted SiO2 particle systems with R0 ≅ 7.7 nm and ρs ≅ 0.5 nm−2 was measured by nanoindentation, Kic0 is the reference value of fracture toughness determined for high molecular weight PS with Mn = 300,000. The fracture toughness increases by about 500% at the CPB → SDPB transition, indicated by transition between differently colored regions. The dotted line is introduced to guide the eye. Insets show transmission electron micrographs of cracks formed during deformation of particle monolayers in CPB and SDPB regime, the direction of the applied stress is indicated by white arrows, and the scale bars are 100 nm.
In conclusion, controlling interfacial interactions in one-component composite materials provides an opportunity to tune Tg and mechanical properties in plastic composites where the competing influence of the stretched and relaxed segments of the grafted brush polymer allows design of particle brush materials with tailored properties.
REFERENCES
(1) Yu, W. H.; Kang, E. T.; Neoh, K. G.; Zhu, S. Journal of Physical Chemistry B 2003, 107, 10198-10205.(2) Matyjaszewski, K. NATO Science Series, II: Mathematics, Physics and Chemistry 2004, 175, 123-134.
(3) Hui, C. M.; Pietrasik, J.; Schmitt, M.; Mahoney, C.; Choi, J.; Bockstaller, M. R.; Matyjaszewski, K. Chem. Mater. 2014, 26, 745-762.
(4) Gao, H.; Matyjaszewski, K. J. Am. Chem. Soc. 2007, 129, 6633-6639.
(5) Huang, J.; Koepsel, R. R.; Murata, H.; Wu, W.; Lee, S. B.; Kowalewski, T.; Russell, A. J.; Matyjaszewski, K. Langmuir 2008, 24, 6785-6795.
(6) Xie, G.; Ding, H.; Daniel, W. F. M.; Wang, Z.; Pietrasik, J.; Sheiko, S. S.; Matyjaszewski, K. Polymer 2016, 98, 481-486.
(7) Ding, H.; Yan, J.; Wang, Z.; Xie, G.; Mahoney, C.; Ferebee, R.; Zhong, M.; Daniel, W. F. M.; Pietrasik, J.; Sheiko, S. S.; Bettinger, C. J.; Bockstaller, M. R.; Matyjaszewski, K. Polymer 2016, 107, 492–502.
(8) Wang, Z.; Lu, Z.; Mahoney, C.; Yan, J.; Ferebee, R.; Luo, D.; Matyjaszewski, K.; Bockstaller, M. R. ACS Applied Materials & Interfaces 2017, 9, 7515-7522.
(9) Khabibullin, A.; Mastan, E.; Matyjaszewski, K.; Zhu, S. Adv. Polym. Sci. 2016, 270, 29-76.
(10) Wang, Z.; Mahoney, C.; Yan, J.; Lu, Z.; Ferebee, R.; Luo, D.; Bockstaller, M. R.; Matyjaszewski, K. Langmuir 2016, 32, 13207-13213.
(11) Pyun, J.; Matyjaszewski, K. Chem. Mater. 2001, 13, 3436-3448.
(12) Wu, L.; Glebe, U.; Boker, A. Polym. Chem. 2015, 6, 5143.
(13) Pyun, J.; Jia, S.; Kowalewski, T.; Patterson, G. D.; Matyjaszewski, K. Macromolecules 2003, 36, 5094-5104.
(14) Ohno, K.; Morinaga, T.; Koh, K.; Tsujii, Y.; Fukuda, T. Macromolecules 2005, 38, 2137-2142.
(15) Von Werne, T.; Patten, T. E. J. Am. Chem. Soc. 1999, 121, 7409-7410.
(16) Yan, J.; Pan, X.; Schmitt, M.; Wang, Z.; Bockstaller, M. R.; Matyjaszewski, K. ACS Macro Lett. 2016, 5, 661-665.
(17) Yan, J.; Pan, X.; Wang, Z.; Zhang, J.; Matyjaszewski, K. Macromolecules 2016, 49, 9283-9286.
(18) Discekici, E. H.; Pester, C. W.; Treat, N. J.; Lawrence, J.; Mattson, K. M.; Narupai, B.; Toumayan, E. P.; Luo, Y.; McGrath, A. J.; Clark, P. G.; Read de Alaniz, J.; Hawker, C. J. ACS Macro Lett. 2016, 5, 258-262.
(19) Mastan, E.; Xi, L.; Zhu, S. Macromol. Theory Simul. 2016, 25, 220−228.
(20) Lin, C. Y.; Coote, M. L.; Petit, A.; Richard, P.; Poli, R.; Matyjaszewski, K. Macromolecules, 2007, 40, 5985-5994.
(21) Ribelli, T. G.; Konkolewicz, D.; Bernhard, S.; Matyjaszewski, K. J. Am. Chem. Soc. 2014, 136, 13303-13312.
(22) Pan, X.; Fang, C.; Fantin, M.; Malhotra, N.; So, W. Y.; Peteanu, L. A.; Isse, A. A.; Gennaro, A.; Liu, P.; Matyjaszewski, K. J. Am. Chem. Soc. 2016, 138, 2411–2425.
(23)
(1) Yu, W. H.; Kang, E. T.; Neoh, K. G.; Zhu, S. Journal of Physical Chemistry B 2003, 107, 10198-10205.
(2) Matyjaszewski, K. NATO Science Series, II: Mathematics, Physics and Chemistry 2004, 175, 123-134.
(3) Hui, C. M.; Pietrasik, J.; Schmitt, M.; Mahoney, C.; Choi, J.; Bockstaller, M. R.; Matyjaszewski, K. Chem. Mater. 2014, 26, 745-762.
(4) Gao, H.; Matyjaszewski, K. J. Am. Chem. Soc. 2007, 129, 6633-6639.
(5) Huang, J.; Koepsel, R. R.; Murata, H.; Wu, W.; Lee, S. B.; Kowalewski, T.; Russell, A. J.; Matyjaszewski, K. Langmuir 2008, 24, 6785-6795.
(6) Xie, G.; Ding, H.; Daniel, W. F. M.; Wang, Z.; Pietrasik, J.; Sheiko, S. S.; Matyjaszewski, K. Polymer 2016, 98, 481-486.
(7) Ding, H.; Yan, J.; Wang, Z.; Xie, G.; Mahoney, C.; Ferebee, R.; Zhong, M.; Daniel, W. F. M.; Pietrasik, J.; Sheiko, S. S.; Bettinger, C. J.; Bockstaller, M. R.; Matyjaszewski, K. Polymer 2016, 107, 492–502.
(8) Wang, Z.; Lu, Z.; Mahoney, C.; Yan, J.; Ferebee, R.; Luo, D.; Matyjaszewski, K.; Bockstaller, M. R. ACS Applied Materials & Interfaces 2017, 9, 7515-7522.
(9) Khabibullin, A.; Mastan, E.; Matyjaszewski, K.; Zhu, S. Adv. Polym. Sci. 2016, 270, 29-76.
(10) Wang, Z.; Mahoney, C.; Yan, J.; Lu, Z.; Ferebee, R.; Luo, D.; Bockstaller, M. R.; Matyjaszewski, K. Langmuir 2016, 32, 13207-13213.
(11) Pyun, J.; Matyjaszewski, K. Chem. Mater. 2001, 13, 3436-3448.
(12) Wu, L.; Glebe, U.; Boker, A. Polym. Chem. 2015, 6, 5143.
(13) Pyun, J.; Jia, S.; Kowalewski, T.; Patterson, G. D.; Matyjaszewski, K. Macromolecules 2003, 36, 5094-5104.
(14) Ohno, K.; Morinaga, T.; Koh, K.; Tsujii, Y.; Fukuda, T. Macromolecules 2005, 38, 2137-2142.
(15) Von Werne, T.; Patten, T. E. J. Am. Chem. Soc. 1999, 121, 7409-7410.
(16) Yan, J.; Pan, X.; Schmitt, M.; Wang, Z.; Bockstaller, M. R.; Matyjaszewski, K. ACS Macro Lett. 2016, 5, 661-665.
(17) Yan, J.; Pan, X.; Wang, Z.; Zhang, J.; Matyjaszewski, K. Macromolecules (Washington, DC, U. S.) 2016, 49, 9283-9286.
(18) Discekici, E. H.; Pester, C. W.; Treat, N. J.; Lawrence, J.; Mattson, K. M.; Narupai, B.; Toumayan, E. P.; Luo, Y.; McGrath, A. J.; Clark, P. G.; Read de Alaniz, J.; Hawker, C. J. ACS Macro Lett. 2016, 5, 258-262.
(19) Mastan, E.; Xi, L.; Zhu, S. Macromol. Theory Simul. 2016, 25, 220−228.
(20) Lin, C. Y.; Coote, M. L.; Petit, A.; Richard, P.; Poli, R.; Matyjaszewski, K. Macromolecules (Washington, DC, U. S.) 2007, 40, 5985-5994.
(21) Ribelli, T. G.; Konkolewicz, D.; Bernhard, S.; Matyjaszewski, K. J. Am. Chem. Soc. 2014, 136, 13303-13312.
(22) Pan, X.; et al., J. Am. Chem. Soc. 2016, 138, 2411–2425.
(23) J. Yan, et al., Chem. Mater. 2017, 29, 4963.
(24) Zhang, J., et al., Polymer 2017, 126, 126-132.]
(25) Pyun, J.; Kowalewski, T.; Matyjaszewski, K. Macromolecular Rapid Communications 2003, 24, 1043-1059.
(26) Tchoul, M. N.; Dalton, M.; Tan, L.-S.; Dong, H.; Hui, C. M.; Matyjaszewski, K.; Vaia, R. A. Polymer 2012, 53, 79-86.
(27) Pietrasik, J.; Hui, C. M.; Chaladaj, W.; Dong, H.; Choi, J.; Jurczak, J.; Bockstaller, M. R.; Matyjaszewski, K. Macromol. Rapid Commun. 2011, 32, 295-301.
(28) Choi, J.; Dong, H.; Matyjaszewski, K.; Bockstaller, M. R. J. Am. Chem. Soc. 2010, 132, 12537-12539.
(29) Bombalski, L.; Min, K.; Dong, H.; Tang, C.; Matyjaszewski, K. Macromolecules 2007, 40, 7429-7432.
(30) Kagawa, Y.; Zetterlund, P. B.; Minami, H.; Okubo, M. Macromolecular Theory and Simulations 2006, 15, 608-613.
(31) Pyun, J.; Jia, S.; Kowalewski, T.; Matyjaszewski, K. Macromolecular Chemistry and Physics 2004, 205, 411-417.
(32) Matyjaszewski, K.; Bombalski, L.; Jakubowski, W.; Min, K.; Spanswick, J.; Tsarevsky, N. In PCT Int. Appl.; (Carnegie Mellon University, USA). WO 2005087819, 2005; p 96 pp.
(33) Min, K.; Yu, S.; Lee, H.-i.; Mueller, L.; Sheiko, S. S.; Matyjaszewski, K. Macromolecules 2007, 40, 6557-6563.
(34) Esteves, A. C. C.; Bombalski, L.; Trindade, T.; Matyjaszewski, K.; Barros-Timmons, A. Small 2007, 3, 1230-1236.
(35) Dong, H.; Zhu, M.; Yoon, J. A.; Gao, H.; Jin, R.; Matyjaszewski, K. J. Am. Chem. Soc. 2008, 130, 12852-12853.
(36) Choi, J.; Hui, C. M.; Pietrasik, J.; Dong, H.; Matyjaszewski, K.; Bockstaller, M. R. Soft Matter 2012, 8, 4072-4082.
(37) Choi, J.; Hui, C. M.; Schmitt, M.; Pietrasik, J.; Margel, S.; Matyjazsewski, K.; Bockstaller, M. R. Langmuir 2013, 29, 6452-6459.