Block Copolymers
Polar thermoplastic elastomers
Reactive surfactants
Molecular assembly of block copolymers
Controlling morphology by PDI
Multi-segmented block copolymers
Block Copolymers
Innumerable block copolymers with novel compositions in each segment have been prepared using CRP procedures, particularly after the initial expansion of the range of copolymerizable monomers that occurred when ATRP was developed. The following discussion focuses on a few examples that show properties significantly different from the properties of materials that could be prepared before 1995. The first class of block copolymers that will be discussed are polar thermoplastic elastomers whose synthesis became much simpler with the development of CRP. A couple of applications building on the inherent elasticity and ability to control monomer composition are described. The second example is the synthesis of block copolymers where each block or segment is selected to accomplish a specific function in the final application.
Polar thermoplastic elastomers:
The major benefit of polar thermoplastic elastomers (TPEs) is that they are oil resistant and recyclable, i.e., it is possible to injection mold the materials and minimize waste by immediately recycling sprues and runners in addition to providing long term recycleability, as required for automotive applications in Europe. The potential market for such materials was estimated to be $2.7 billion/yr market in 2007.(1) Polar TPEs are materials that resistant to hydrocarbon solvents (e.g., fuels). TPEs provide the softness, flexibility and resilience of elastomers with the processability of thermoplastics.
The most common examples of commercial TPEs are poly(styrene-b-butadiene-b-styrene) (e.g., Kraton®) and polyurethane-polyether and polyester-polyether multiblock copolymers. However, the materials that can now be prepared by CRP provide several additional benefits including greater versatility in monomer choice, which provides materials where the Tg and phylicity of hard and soft block can be selectively chosen, or tailored to attain specific properties required for the application. Additional functionality can be incorporated into one or both blocks. Since multi-functional initiators are easily prepared other architectures can be considered (e.g., star-blocks, grafts, brushes with block side-chains, etc.) since polymer topology also affects properties.(2)
The desire to prepare well defined TPEs led to the development of "halogen exchange". The use of "halogen exchange" in a polymerization increases the rate of initiation in the critical cross propagation chain extension step, from the added, or in situ prepared macroinitiator,(3) compared to the rate of propagation of the second monomer and is required for the preparation of well defined P(MMA-BA-MMA) block copolymers prepared using a difunctional initiator.(4)
The phase state of a series of poly(n-octadecyl methacrylate)-b-poly(t-Bu acrylate)-b-poly(n-octadecyl methacrylate) (pODMA-b-ptBA-b-pODMA) triblock copolymers has been investigated in bulk and on surfaces using small-angle X-ray scattering and AFM respectively.(5) The mean-field theory was employed to construct the bulk phase diagram. Excellent agreement was found between the bulk and surface morphologies as well as for the domain spacing (domain spacing scaled as d ~ N0.64), suggesting that the strong polymer-polymer interactions in bulk are also the dominant interactions on surfaces.
In order to avoid the necessity of "halogen exchange" an all methacrylate block copolymer can be prepared.(6) For example the triblock copolymer poly(Me methacrylate-b-lauryl methacrylate-b-Me methacrylate) has been synthesized by a two stage ATRP in bulk at near room temperature (ca. 35 0C). A CuCl/pentamethyldiethylenetriamine (PMDETA)/tricaprylylmethylammonium chloride (Aliquat 336) complex was used as the catalyst and 1,2-bis (bromoisobutyryloxy)ethane (BIBE) as the initiator for the polymerization of LMA in the first stage. The same catalyst was also used for the polymerization of MMA in the second stage. The dynamic mechanical thermal analysis of a sample with the middle block Mn = 82,000 and each end block Mn = 14,500 showed features typical of a thermoplastic elastomer.
As noted above the properties of a TPE can be readily modified in a CRP by incorporation of copolymer blocks into the design of the material. However, rather than discuss further such a linear polar TPE macromolecule we will discuss some recently published data on the properties of PBA-b-PAN 3-arm star block copolymers.(7,8) These materials are polar TPE's with easily adjustable properties, based on composition, that retain their useful properties over a temperature range: -50 to +250 0C!
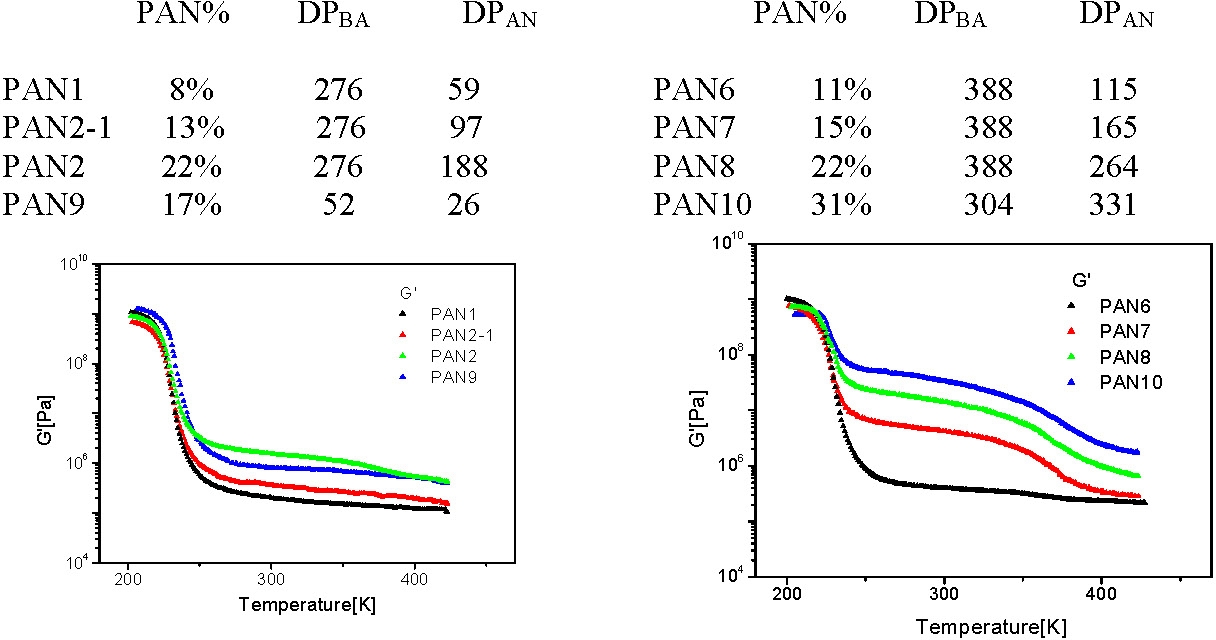
The compositions of the star block copolymers discussed below are given in the following tables.
Observations:
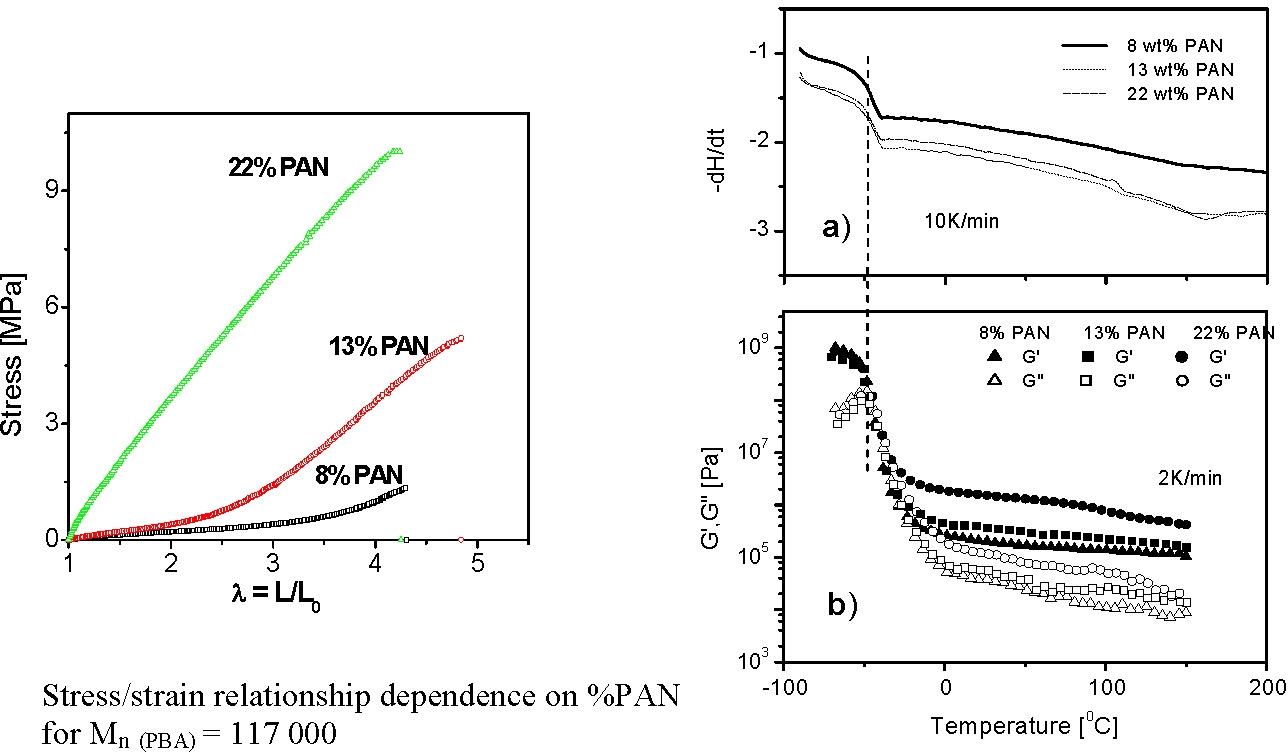
- all samples are glassy below the Tg of pBA, (~227K), with G´~ GPa;
- above the Tg of pBA the materials remain elastic with a G´ plateau extending up to the softening temperature of pAN (~370K).
- The height of the plateau is dependent on the pAN content of the segmented copolymer: G´~0.1 MPa when pAN fraction < 0.1 and G´~100 MPa when pAN fraction is 0.3.
- Above the softening temperature of the pAN block the material remains elastic with a lower G´ plateau (0.1 - 1 MPa) extending up to temperature of ~550K.
- This behavior probably indicates some chemical effects such as chemical crosslinking take place above the melting temperature.
(Note similar materials are used as precursors for nano-structured carbons.)(7)
In the following figures the mechanical spectrum resulting from quasi-static stress-strain cycles with successively increasing maximum strain are shown in different colors. The PAN1 sample exhibits nearly ideal elastic behavior: no residual strains after unloading. In the case of samples with slightly higher mole fractions of PAN the elasto-plastic behavior shows large residual strains after unloading the plastic deformation which is probably associated with fragmentation of the pAN cylindrical domains.
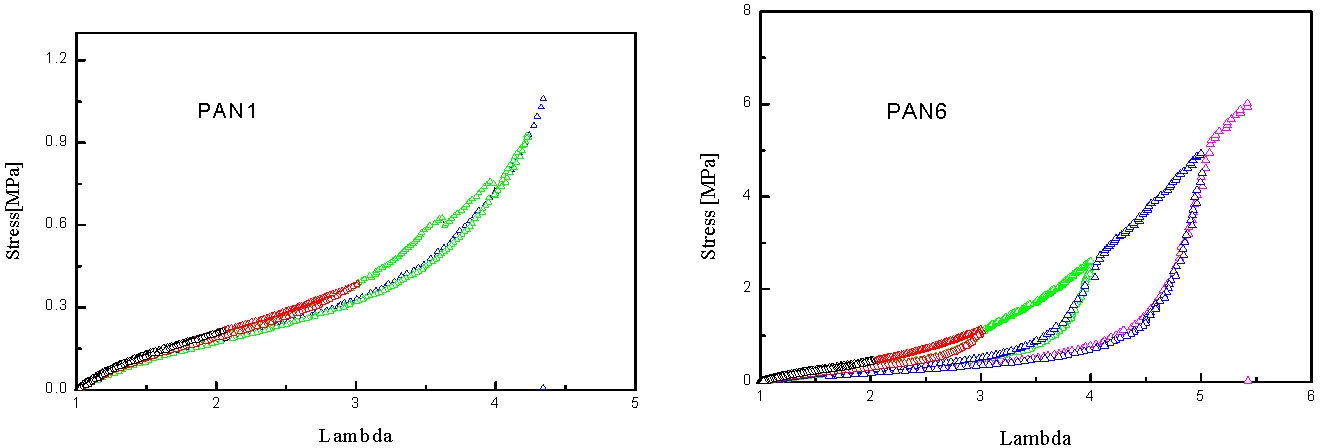
Under quasi-static stretching the modulus increases with pAN content and the draw ratio at break seems to increase with the length of pBA segment and be reduced with increasing pAN content.
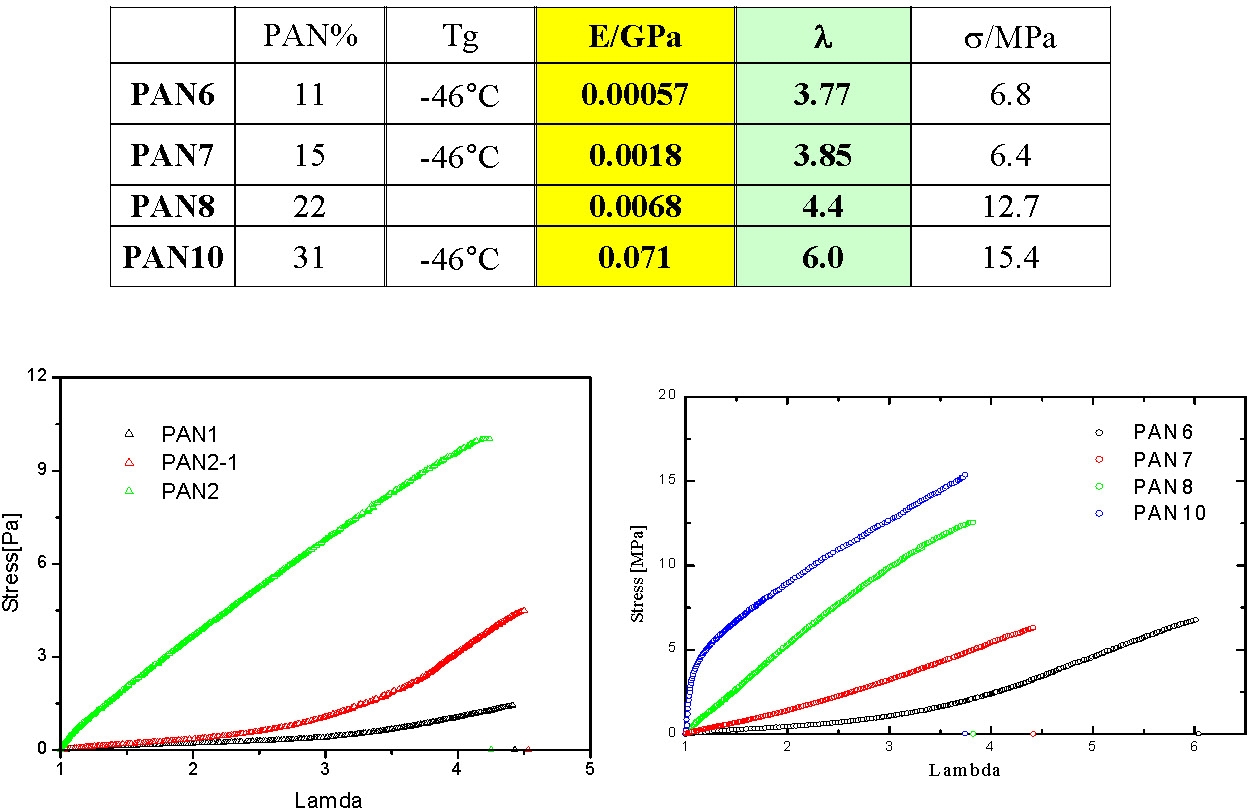
High temperature annealing indicated a hardening of the annealed samples which probably results from improved morphological order. A sample annealed above 300°C remains elastic at temperatures above the softening point of pAN.
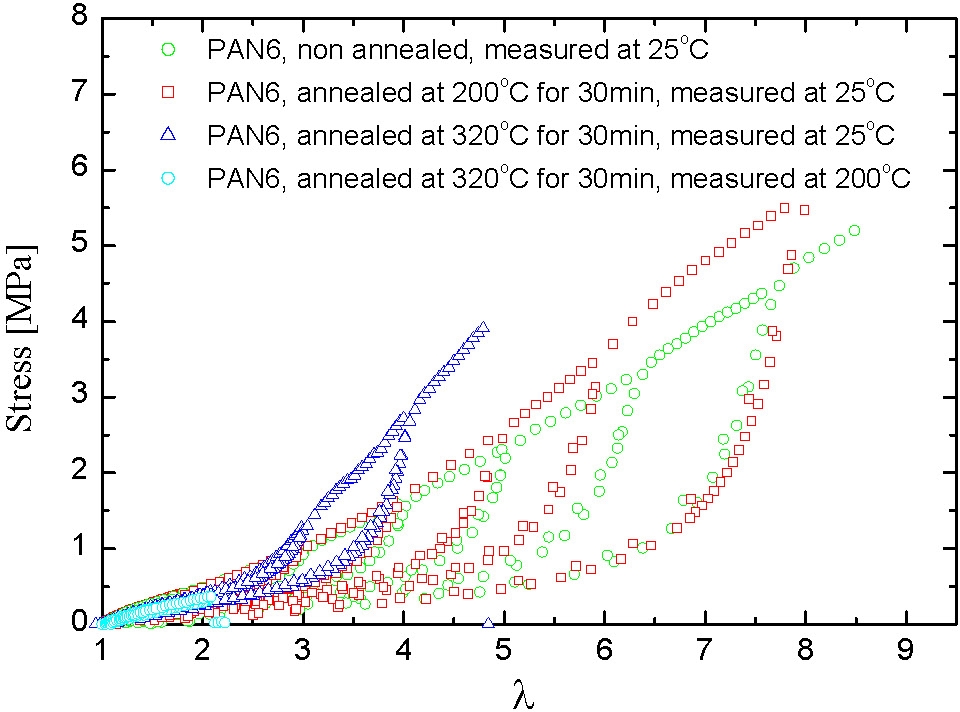
This example shows how the properties of a thermoplastic elastomer can be readily modified by changing the volume fraction of the A-blocks in the final product.
Thermoplastic elastomers can be synthesized in a one-pot process using ARGET. PS-PEA-PS and PMMA-PBA-PMMA block copolymers were made in such a way,(3) and mechanical testing should be done soon and this will be published.
While applications based on the bulk physical properties of thermoplastic elastomers are almost the "classic" markets for segmented triblock copolymers other properties inherent in the chosen monomer units can be selected to target other applications. Acrylate-based block copolymers were evaluated as drug delivery matrixes for the controlled release of paclitaxel from coronary stents. (9) The polymers were multiblock copolymers consisting of poly(Bu acrylate) or poly(lauryl acrylate) soft blocks and hard blocks composed of poly(Me methacrylate), poly(isobornyl acrylate), or poly(styrene) homo- or copolymers. Depending on the ratio of hard to soft blocks in the copolymers, coating formulations were produced that possessed variable elastomeric properties, resulting in stent coatings that maintained their integrity when assessed by SEM imaging of overexpanded stents. In vitro paclitaxel release kinetics from coronary stents coated with these copolymers typically showed an early burst of paclitaxel followed by sustained release behavior, which permitted the elution of the majority of the paclitaxel over a 10-day time period.
Another high value application was developed by IBM when they developed a block copolymer for use in chip manufacturing:
http://www.usatoday.com/tech/news/techinnovations/2007-05-03-ibm-chip_N.htm
The block copolymer undergoes self-assembly when applied on top of a substrate and is treated to form tiny holes in the matrix by selective depolymerization of one block. The chip is then exposed to a gas that seeps through the material as if it were a stencil, etching away the underlying glass to form small holes in the top surface, and larger, continuous gaps between the wires. Another layer of glass is applied in a vacuum chamber. Because the holes in the topmost existing glass layer are small, the newly applied layer of glass doesn't seep into the underlying cavities. Instead, it seals them off, with a vacuum inside. The technique was invented at IBM's Almaden Research Center in San Jose, Calif., and the T.J. Watson Research Center in Yorktown, N.Y and was adapted for commercial use by the University at Albany and IBM's Semiconductor Research and Development Center in East Fishkill, N.Y.
REFERENCES
(1) Global Information Incorporated Press release 12 19 03.
(2) Spanswick, J.; Matyjaszewski, K. TPEs by CRP Chapter 14, 3rd Edition ed., 2004.
(3) Jakubowski, W.; Spanswick, J.; Mueller, L.; Matyjaszewski, K. In WIPO; CARNEGIE MELLON UNIVERSITY: WO/2007/075817, 2007.
(4) Matyjaszewski, K.; Shipp, D. A.; McMurtry, G. P.; Gaynor, S. G.; Pakula, T. J. Polym. Sci., Part A: Polym. Chem. 2000, 38, 2023-2031.
(5) Wu, W.; Huang, J.; Jia, S.; Kowalewski, T.; Matyjaszewski, K.; Pakula, T.; Gitsas, A.; Floudas, G. Langmuir 2005, 21, 9721-9727.
(6) Chatterjee, D. P.; Mandal, B. M. Macromolecular Symposia 2006, 240, 224-231.
(7) Dufour, B.; Tang, C.; Koynov, K.; Zhang, Y.; Pakula, T.; Matyjaszewski, K. Macromolecules 2008, 41, 2451-2458.
(8) Dufour, B.; Koynov, K.; Pakula, T.; Matyjaszewski, K. Macromol. Chem. Phys. 2008, 209, 1686-1693.
(9) Richard, R. E.; Schwarz, M.; Ranade, S.; Chan, A. K.; Matyjaszewski, K.; Sumerlin, B. Biomacromolecules 2005, 6, 3410-3418.