"Reverse" ATRP
In the initially developed "reverse" ATRP initiation procedure transition metal complexes in the higher oxidation state (e.g. CuII complex) are added to the reaction and the ATRP initiator (I-X or I-P1-X) and the activator are generated in situ by reactions triggered by decomposition of a conventional free radical initiator.(1-5)
In a "reverse" ATRP the components that are added to the initial system are less sensitive to air, therefore the catalyst precursors are easier to handle, particularly with active catalyst complexes, and the procedure was thought to be more compatible with industrial scale processes since standard free radical initiators are employed. This means that the initiation step does not proceed by activation of an alkyl halide with a Mtn/L catalyst, but rather by thermal decomposition of a conventional free radical initiator, such as AIBN. Once radicals are generated, they either react immediately with the higher oxidation state transition-metal complex to form the reduced transition-metal species and a dormant species, I · + X-Mtn+1/L forming I-X + Mtn/L, or they react with monomer to form a propagating radical, I-P1·, which is then quickly deactivated by reaction with the added X-Mtn+1/L to form Mtn/L and a dormant species, I-P1-X.
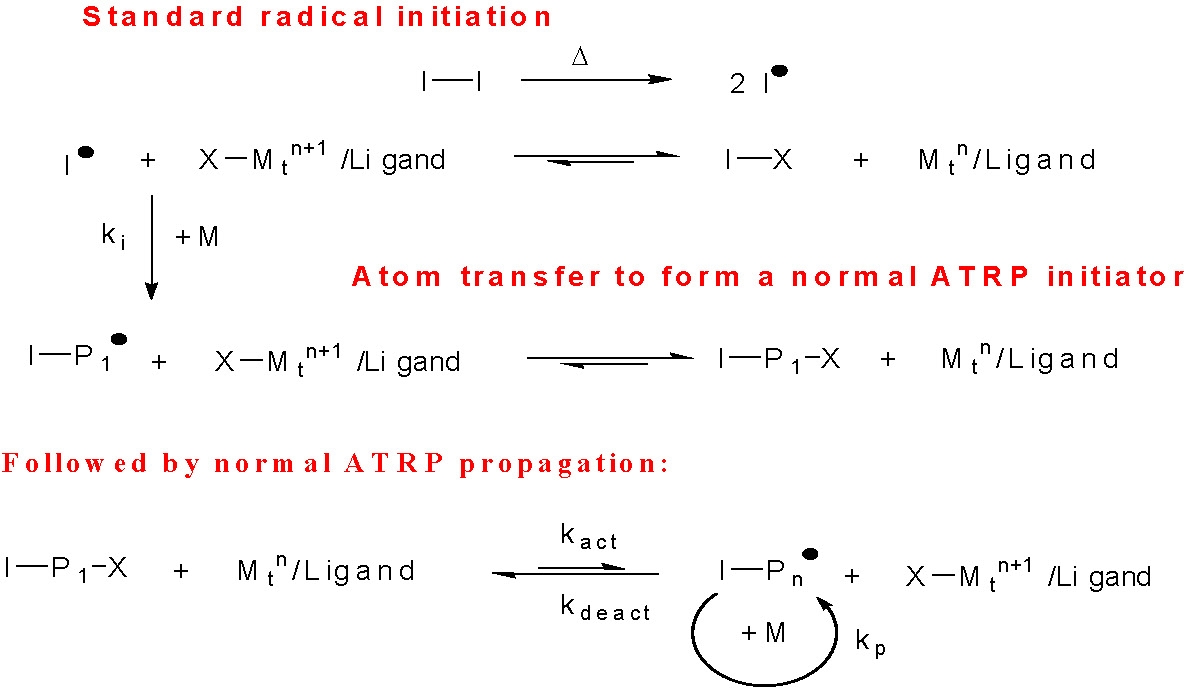
In subsequent steps, the initially formed reduced transition-metal species, Mtn/L, reacts with the newly formed halogen terminated chains, I-X or I-P1-X, as in a normal ATRP initiation/propagation process.
As noted above, the advantages of reverse ATRP include starting with the more stable transition metal complex, which is particularly useful when one wants to use more active catalyst complexes that are easily oxidized,(6) or for miniemulsion systems,(6,7) for the preparation of a range of linear copolymers with good ω-chain end functionality.
The disadvantages of the initial "reverse" ATRP procedures are that it limits the terminal functionality remaining on the initiator residue (α-functionality) to that present as an additional functional group on the "I" residue of the standard free radical initiator, and limits the topology of the polymers that can be prepared. Only linear (co)polymers can easily be prepared. Furthermore the molecular weight of the final copolymer cannot be independently adjusted irrespective of the activity of the transition metal complex, since the transferable atom or group on the growing polymer chain end is introduced as a ligand on the added catalyst.
[M]0/[CuIIX2-ligand]0/[I·]0 = DP target/I-I
Consequently a relatively high amount of catalyst (CuII-ligand) is required:
[CuII-ligand]0/[I - I]0 = 1 - 1.5
![]()
REFERENCES
(1) Tang, W.; Matyjaszewski, K. Macromolecules 2007, 40, 1858-1863.
(2) Fischer, H. J. Polym. Sci., Part A: Polym. Chem. 1999, 37, 1885-1901.
(3) Wang, J.-S.; Matyjaszewski, K. Macromolecules 1995, 28, 7572-7573.
(4) Xia, J.; Matyjaszewski, K. Macromolecules 1997, 30, 7697-7700.
(5) Moineau, G.; Dubois, P.; Jerome, R.; Senninger, T.; Teyssie, P. Macromolecules 1998, 31, 545-547.
(6) Woodworth, B. E.; Metzner, Z.; Matyjaszewski, K. Macromolecules 1998, 31, 7999-8004.
(7) Li, M.; Matyjaszewski, K. Macromolecules 2003, 36, 6028-6035.