Photoinduced ATRP, (π-ATRP)
(in the absence of Photoinitiators)(1)
Prior work on photoinduced initiation of an ATRP employed reverse ATRP or SR&NI procedures in conjunction with a standard photoinitiator. Yagci et al. (2-3) discussed the photochemical generation of the activator from the added CuIIX/L complex in the presence of methanol and subsequent reaction of the activator with the added alkyl halide. They employed a high concentrations of catalyst while Mosnacek used UV light to activate a reaction with lower concentrations of catalyst.(4)

Our interest in photoinitiation of an ATRP arose out of a desire to expand the possibility of photoinduced reduction of CuII halide complexes to visible light and determine if it was possible that excited state CuII species could undergo redox processes that are inaccessible from the ground state. The microscopic control of these photochemical reactions, due to the simple triggering by visible light, was envisioned to be an attractive feature for building useful materials with complex molecular architectures.
The mechanism of the photoinduced ATRP was studied using narrow bandwidth light emitting diodes (LEDs). This avoids complications due to absorption at two or more different wavelengths. The photoinduced ATRP was also used to make block copolymers, and the reaction was performed in water. The reaction was also conducted in the presence of sunlight.(1) LEDs were selected as the light source since they are; inexpensive, efficient, exhibit a long lifetime and are available in various wavelengths with narrow emission range. Photo-reactors were created by running a 5 m strip of LEDs inside a circular chamber.

The lights emitted in: violet (392 ± 7 nm), blue (450 ± 10 nm) and red (631 ± 9 nm)
The reactors were used for the polymerization of methyl methacrylate, oligo(ethylene oxide) monomethyl ether methacrylate (molecular weight 300) (OEOMA), ethyl acrylate (EA) and methyl acrylate (MA) with ethyl
a--bromoisobutyrate (EBiB) as the initiator for acrylates and ethyl α-bromophenylacetate (EBPA) for methacrylates with 100 ppm of CuBr2 complexes with tris(2-pyridylmethyl) amine (TPMA), N,N,N’,N”,N”-pentamethyldiethylenetriamine (PMDETA) and with tris((4-methoxy-3,5-dimethylpyridin-2-yl)methyl)amine (TPMA*) as ligands. UV/Vis/NIR spectroscopy was used to characterize the complexes. The CuII complexes absorb very strongly in the UV with some absorption in the violet region, weak absorption in the blue region, and weak absorption in the red region.
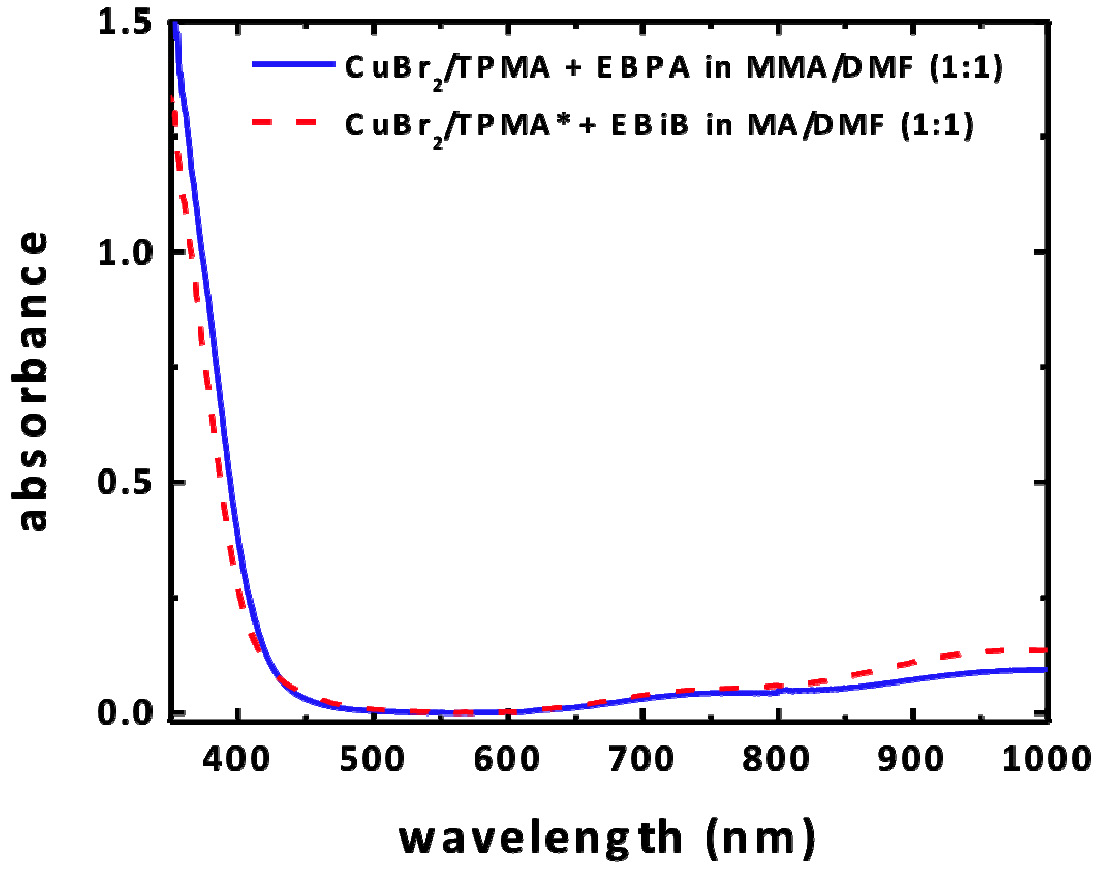
Absorbance measured for reactionwith initial concentration of reagents [M]0:[I]0:[CuBr2]0:[L]0 = 300:1:0.03:0.135, [M] = 4.7/5.5 M in DMF
When using different wavelength for the LEDs, the rates of polymerization of MMA followed the absorbance. The reaction was fastest with violet light, followed by blue light and no polymerization occurred with red light, and the control was good with both violet and blue light. The rate of polymerization did not change significantly in solvents of differing polarity. Blank experiments conducted in the absence of initiator or catalyst progressed at a lower rate, generating polymers with high molecular weight and broad dispersity, which provided insight into the mechanism.
The results reported in the paper are best explained by the photoreduction of the X-CuII/L deactivator complex to generate CuI/L and a halogen radical. The halogen radical can react with monomer to initiate a growing polymer chain that is subject to the ATRP equilibrium. When alkyl halide initiators are added these new chain represent a very small fraction of growing chains. The procedure can be considered a hybrid of ICAR (5) and ARGET ATRP.
The reaction does not progress in the absence of light and reactions with intermittent on-off switching of light progress in a well controlled manner with molecular weight close to the theoretical values and narrow dispersity.
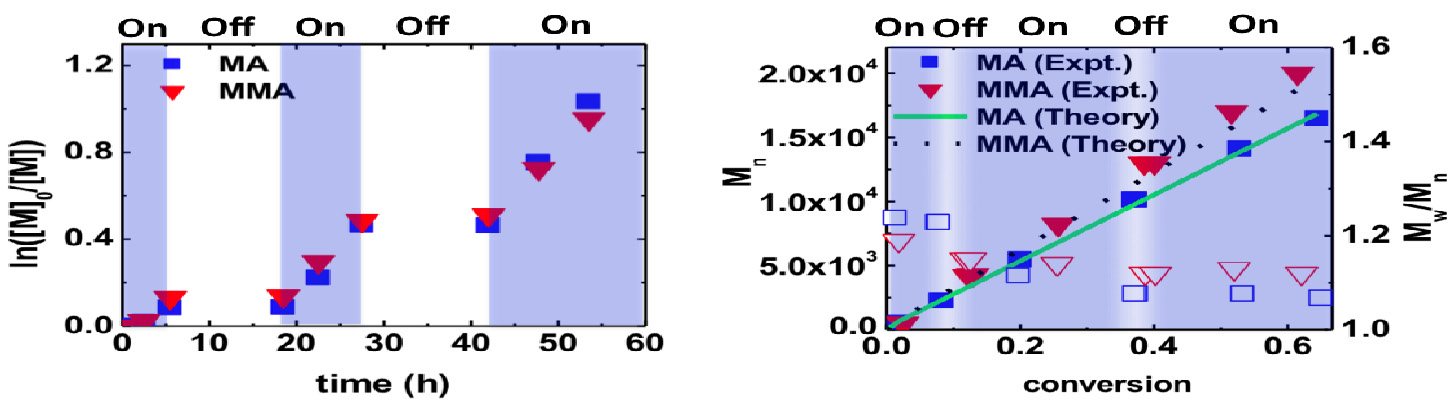
One key advantage of ATRP is that this technique can be used to form well defined block copolymers, as seen by the chain extension of a PMMA macroinitiator formed using this photoinduced activation procedure (π-ATRP), which itself was chain extended with ethyl acrylate providing polymers of higher MW with minimal tailing in the GPC traces.
The procedure was extended to polymerize oligo(ethylene oxide) monomethyl ether methacrylate in water (67%), in the presence of an added halide salt to promote the formation of the deactivator.(1) The polymerization was conducted at room temperature in the presence of violet radiation. Conversion reached 60% in 11.5 h forming a polymer of 65,000 MW with Mw/Mn = 1.34
Theoretically the least expensive light source is the sun, and the photoinduced ATRP of MA and MMA with sunlight is fast, yet still well controlled.
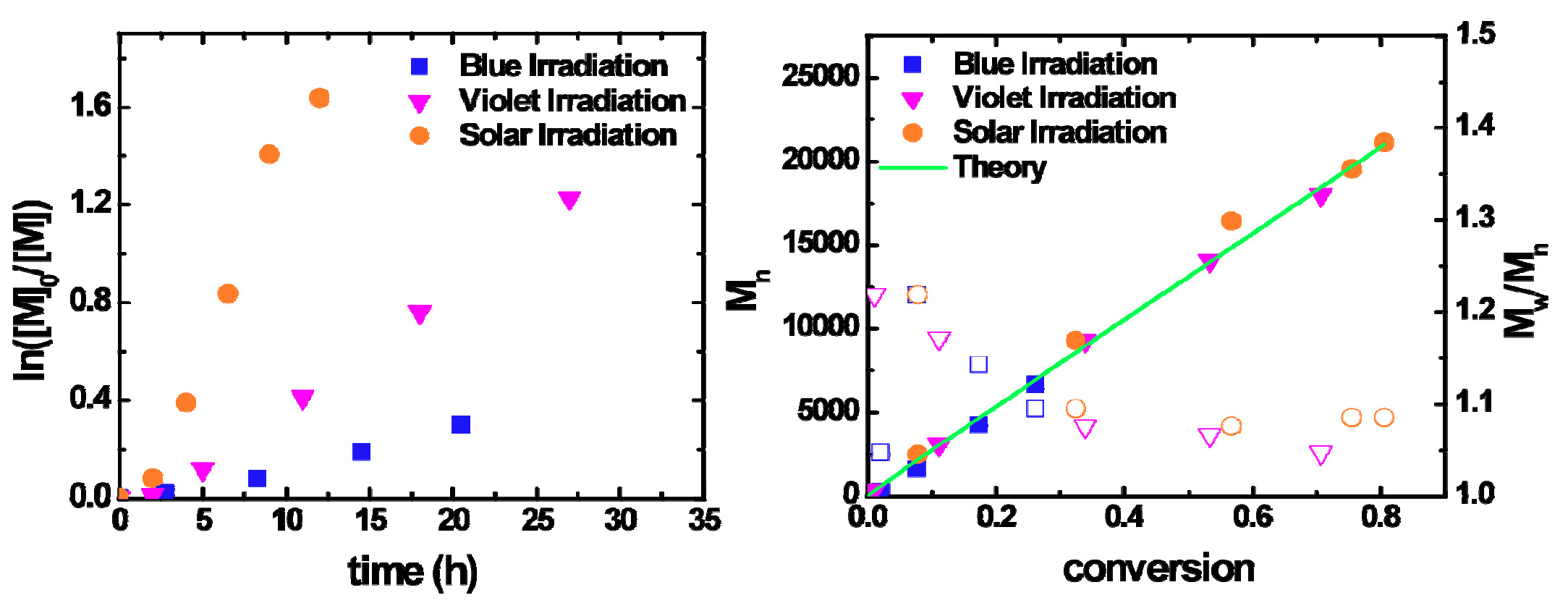
[M]0:[I]0:[CuBr2]0:[L]0 = 300:1:0.03:0.135, [M] = 5.5 M in DMF.
In summary photochemical ATRP is possible under mild reaction conditions with LED irradiation or sunlight, and importantly no unnecessary byproducts are formed. The procedure can be employed to create block copolymers, can be performed in water and the reaction can be modulated by controlling light intensity and wavelength.
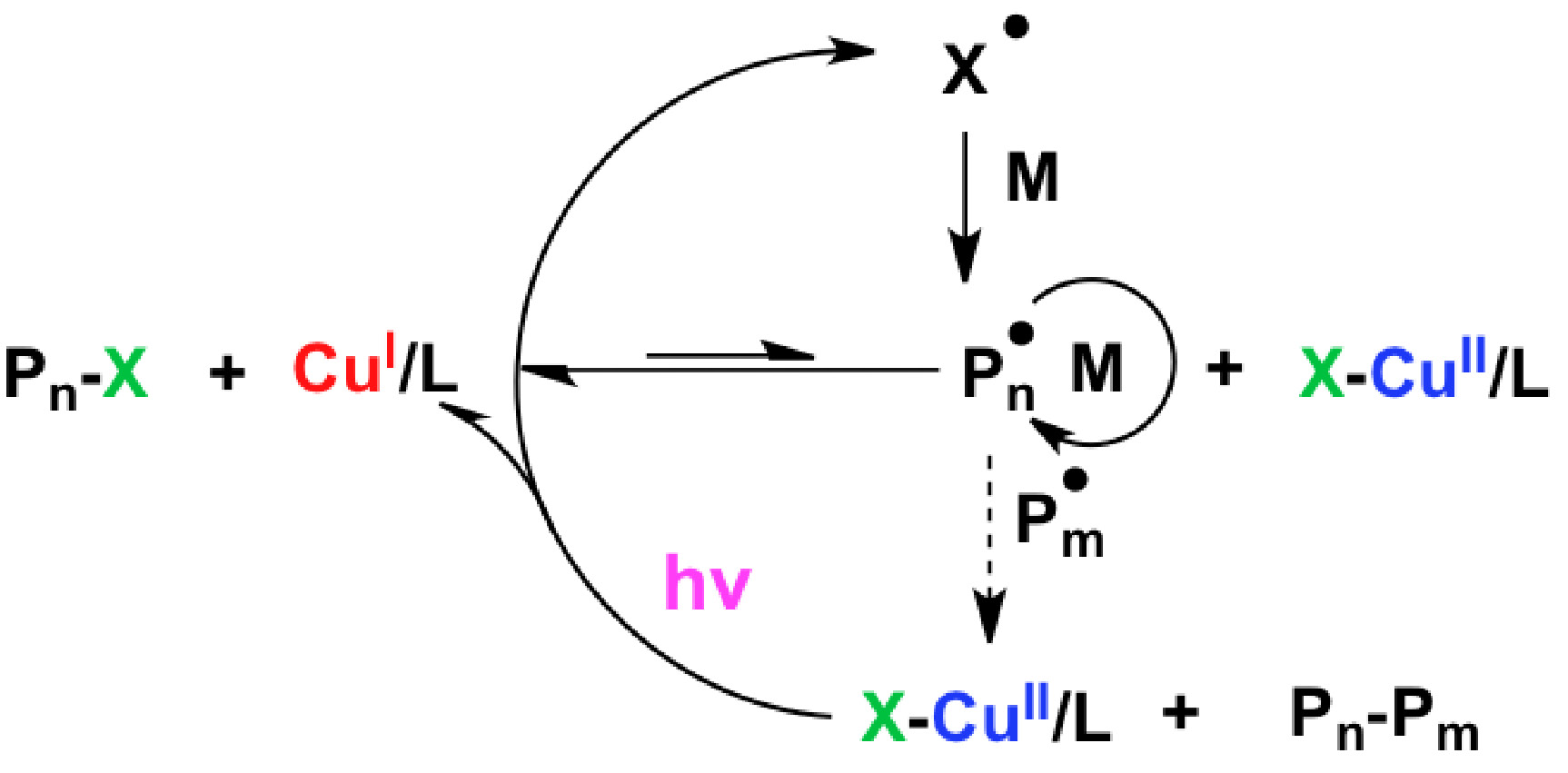
Subsequent work sought to determine “How are Radicals (Re)generated in Photochemical ATRP”. (6) The polymerization mechanism of photochemical mediated Cu-based ATRP was extensively investigated using both experimental and kinetic modeling techniques. As shown in the following schematic there are several distinct pathways that can lead to photochemical (re)generation of CuI activator species or formation of radicals,
Proposed activator (re)generation pathways in PhotoATRP.
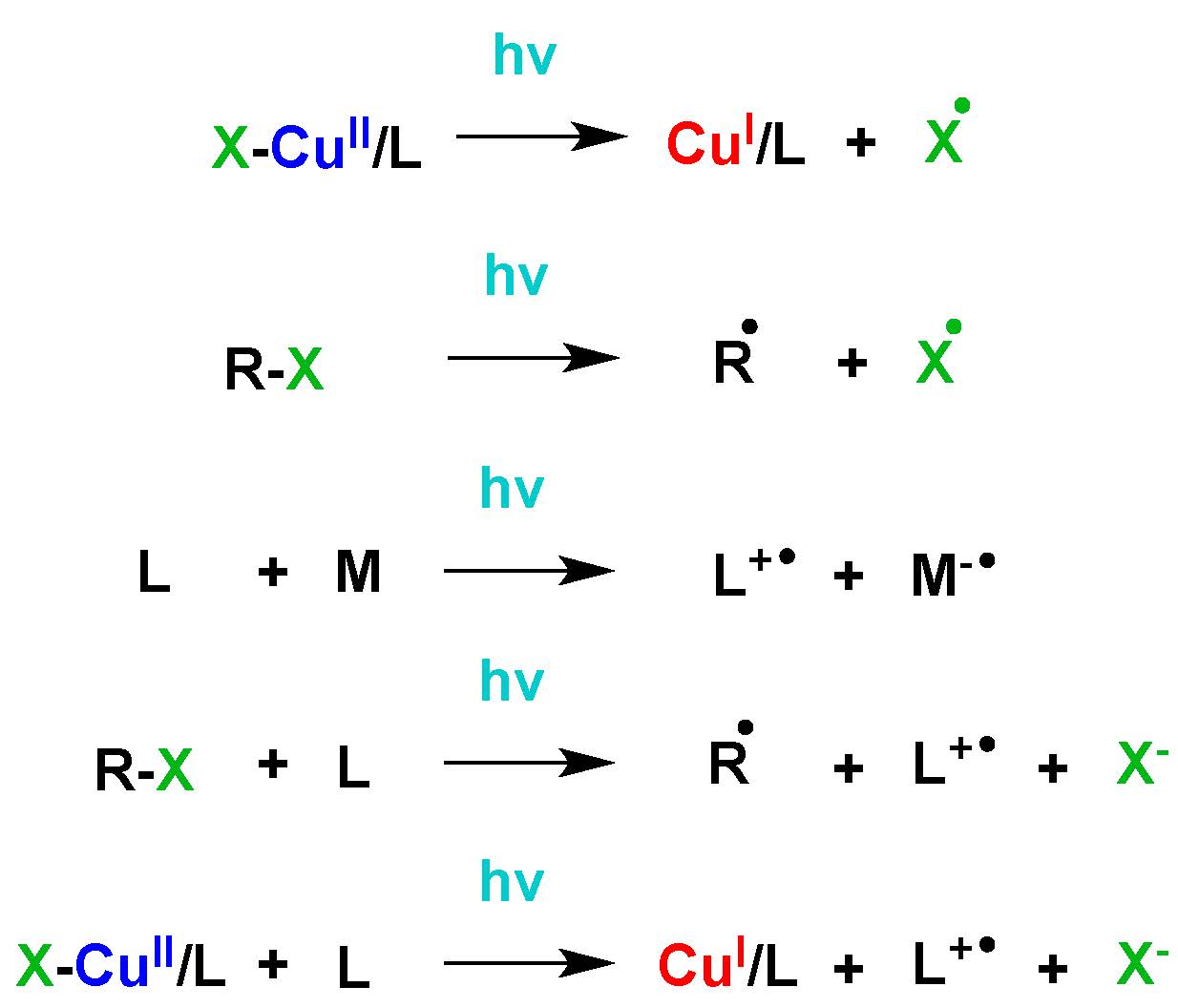
The upper pathway is direct reduction of XCuII/L, while the middle three are the generation of radicals by the reactions of an alkyl halide and/or a ligand, and bottom is the photochemical reduction of CuII by an electron donor.
These potential (re)generation pathways include direct photochemical reduction of the CuII complexes by excess free amine moieties and unimolecular reduction of the CuII complex, which could be considered to be a hybrid between activators regenerated by electron-transfer (ARGET) or initiator for continuous activator regeneration ICAR) ATRP processes, since CuII is reduced in the presence of light by electron transfer as in ARGET while giving a halogen radical that can initiate new chain, as in ICAR ATRP. An alternative radical (re)generation mechanism has the photo-excited alkyl halide, ligand, or their combined interaction, generating a radical species which can react with monomer in a photochemical mechanism akin to ICAR ATRP, as shown in lines 2-4 of the schematic. These photochemical radical generation processes are similar to initiators for continuous activator regeneration (ICAR) ATRP processes.
In the case of the alkyl halide, homolytic cleavage of the carbon-halogen bond is anticipated as shown in line 2 of the schematic. (7) In the case of the photochemical interaction involving the ligand, the nitrogen centered radical cation is expected to be generated (8) and in order conserve both charge and spin, a second molecule must accept this electron. This alternative molecule can be an electron poor alkene, such as a (meth)acrylate moiety as shown in the third line of the scheme. (8) Alternatively, the ligand and alkyl halide can photochemically generate the nitrogen centered radical cation, an alkyl radical and a halide anion, as shown in line 4. A final possibility is that the CuII complexes in the excited state can react with electron donating species, e.g. an amine based ligand, reducing the CuII species and generating a radical cation species from the ligand, as shown in line 5. In all cases where the nitrogen centered radical cation is generated, it rapidly undergoes proton transfer, giving a protonated amine and a carbon centered radical. It should be noted that all of these photochemical radical (re)generation pathways are parallel pathways.
A series of model experiments, ATRP reactions and kinetic simulations, were performed to evaluate the contribution of these reactions to the photochemical ATRP process (6) using 392 nm radiation. The results of these studies clearly indicated that the dominant radical (re)generation reaction is the photochemical reduction of CuII complexes by free amines moieties, from amine containing ligands. The unimolecular reduction of the CuII deactivator complex is not significant and virtually no polymerization occurs in the absence of free ligand. (9) Escentially this is an ARGET ATRP process with the amine being oxidized to a radical cation which can initiate a new chain after proton transfer. However, there is some contribution from ICAR ATRP reactions involving the interaction of alkyl halides and ligand, ligand with monomer, and the photochemical cleavage of the alkyl halide. Nevertheless the predominant mechanism of photochemical mediated ATRP is consistent with a photochemical ARGET ATRP reaction dominating the radical (re)generation process
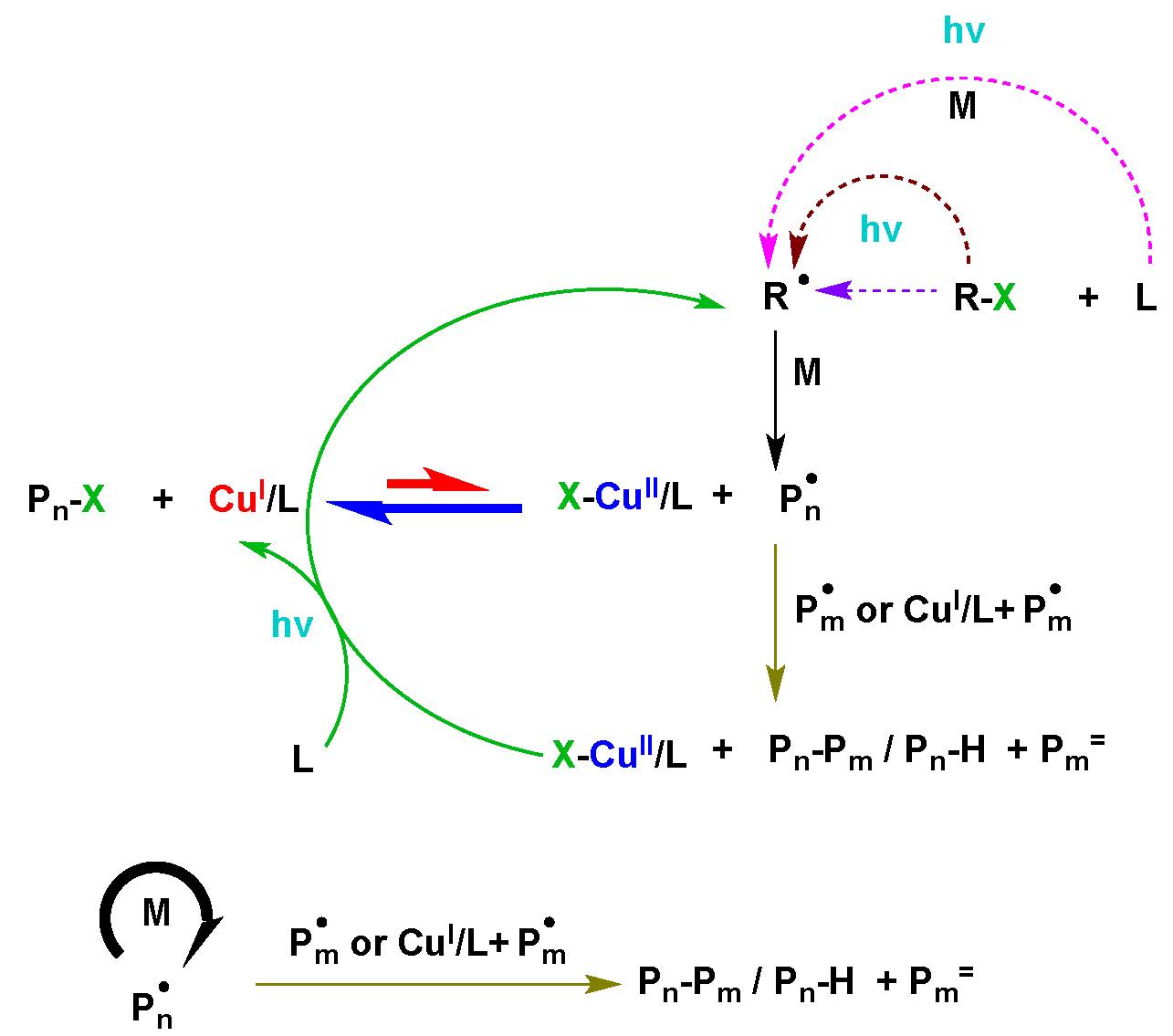
In the above summary of all potential contributing reactions to a photochemical ATRP, reactions with the highest rate are in bold arrows while reactions that dictate the rate are thin solid lines and reactions with low contributions to the mechanism are dashed lines.
(1) Konkolewicz, D.; Schroder, K.; Buback, J.; Bernhard, S.; Matyjaszewski, K. ACS Macro Letters 2012, 1, 1219-1223.
(2) Tasdelen, M. A.; Uygun, M.; Yagci, Y. Macromolecular Chemistry and Physics 2011, 212, 2036–2042.
(3) Tasdelen, M. A.; Ciftci, M.; Uygun, M.; Yagci, Y. ACS Symp. Ser. 2012, 1100, 59-72.
(4) Mosnacek, J.; Ilcikova, M. Macromolecules 2012, 45, 5859-5865.
(5) Konkolewicz, D.; Magenau, A. J. D.; Averick, S. E.; Simakova, A.; He, H.; Matyjaszewski, K. Macromolecules 2012, 45, 4461-4468.
(6) Ribelli, T. G.; Konkolewicz, D.; Bernhard, S.; Matyjaszewski, K. J. Am. Chem. Soc. 2014, 136, 13303-13312.
(7) Klán, P.; Wirz, J. Photochemistry of Organic Componds: From Concepts to Practice; John Wiley & Sons, 2009.
(8) Lewis, F. D.; Crompton, E. M. SET Addition of Amines to Alkene, 2nd ed.: CRC Press: Boca Raton, 2010.
(9) Anastasaki, A.; Nikolaou, V.; Zhang, Q.; Burns, J.; Samanta, S. R.; Waldron, C.; Haddleton, A. J.; McHale, R.; Fox, D.; Percec, V.; Wilson, P.; Haddleton, D. M. J. Am. Chem. Soc. 2014, 136, 1141-1149.
Photoinduced ATRP with ppm Levels of Catalyst
Two advances were made in the above photoinduced ATRP procedures in 2015, each employing only ppm levels of transition metal catalyst; one was conducting the reaction in aqueous media (1) and the other conducting an iron based photoinduced ATRP in the absence of additional ligands, reducing agents or radical initiators. (2)
Aqueous Photoinduced ATRP with ppm levels of copper
Conducting an ATRP in homogeneous aqueous media is challenging due to the high value for KATRP, partial dissociation of the halide ion from the deactivator complex and CuI disproportionation. (3, 4) However recent papers from the group have disclosed successful low ppm level aqueous ATRP for ICAR, (5) ARGET, (6) and SARA ATRP, (7) each providing good control over the rate of polymerization while generating polymers with narrow MWD. However since each of these procedures introduced additional reactants into the reaction it was desirable to determine if a “cleaner” procedure could be developed. A series of photoinduced aqueous ATRP reactions were conducted with different concentrations of copper in the presence of TPMA as ligand (1) and it was determined that catalyst concentrations as low as 22 ppm, with respect to monomer, under irradiation with visible light, 392 nm at 0.9 mWcm2, provided a well-controlled polymerization.
The effect of catalyst concentration, added salt, and targeted degree of polymerization were examined and, in addition to confirming that the polymerization could be directly regulated by external photo-stimulation, it was confirmed that the polymers retained chain end functionality by clean chain extension to form block copolymers.
OEOMA500 was selected as the water soluble monomer for studies of aqueous photo-ATRP utilizing a CuBr2/TPMA catalyst complex using poly(ethylene oxide)-bromophenyacetate as a macroinitiator under visible light irradiation at room temperature. TPMA was selected as the ligand since it forms a stable CuI complex without any significant disproportionation in water, and the air-stable CuII deactivator could be efficiently reduced to the CuI activator under photochemical conditions. (8) In order to reduce the possibility of dissociation of the halogen atom from the CuBr2/TPMA complex, and shift the equilibrium towards the Br-CuIIBr/TPMA deactivator complex, an excess of halide salt was added to the reaction to promote efficient deactivation. The rate of polymerization decreased after the addition of 5 mM NaBr and control over the molecular weight and dispersity improved. The best result currently attained by a photoinduced ATRP was provided using 22 ppm of Cu catalyst with 30 mM salt. The polymerization reached 55% conversion after 4 h forming a polymer with Mn = 103,000 and Mw/Mn = 1.17. Different degrees of polymerization were targeted and a different monomer, OEOMA330, was also examined. All polymerizations provided well-defined polymers with low values for Mw/Mn, between 1.10 and 1.18 and molecular weight agreeing well with theoretical values.
Photoinduced Iron-based ATRP
Why iron? Iron is the most common element on earth by mass, it is the 4th most common element in the Earth's crust, and is the most abundant metal and the most common metal in industrial use in addition to being relatively inexpensive, $1 or less per kg.
FeBr2 ATRP of MMA has been carried out in a series of polar solvents: NMP, DMF, MeCN and DMSO with a ratio of [MMA]:[EBrPA]:[FeBr2] = 100:1:1, MMA/solvent = 2/1 (v/v), at 60oC with the following results.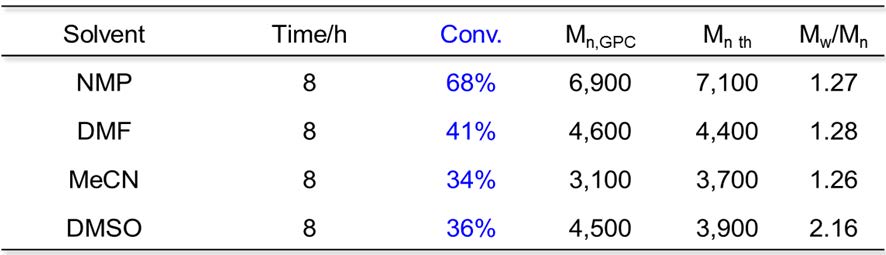
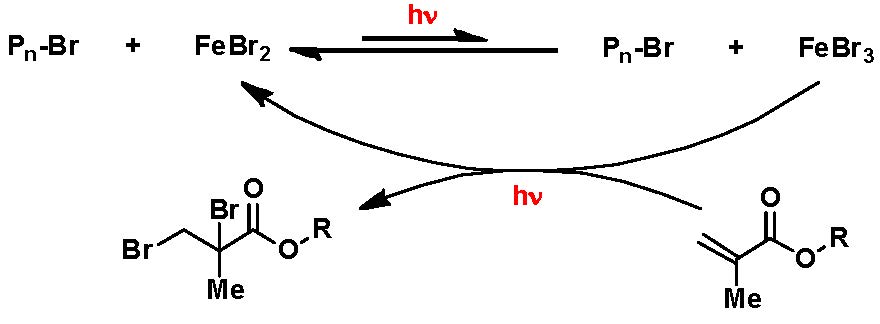
In addition, FeBr3 ATRP of MMA was carried out in the presence of ligands but in absence of a separate reducing agent or radical initiator(9, 10) The question then became can we conduct an FeBr3 based ATRP in absence of ligand, reducing agent or radical initiator solely induced by photochemical activation? Indeed under the following conditions: MMA/EBPA/FeBr3 = 100/1/1, 33.3% MeCN, 4.9 mW/cm2 UV hv, at room temperature, after a 7 h induction period required for the reduction of FeIII, a relatively slow controlled polymerization was observed with a linear increase in MW with conversion, reaching 60% conversion after 26 h, forming polymer with low Mw/Mn.
The reaction was also conducted in the absence of an added ATRP initiator. Since only FeBr3 was used, no FeBr2 added, the successful polymerization indicated that some reduction of FeBr3 must have occurred in order to activate the reaction. The most likely reduction reaction is that FeBr3 was reduced by MMA to form FeBr2 and Br-MMA-Br, as shown in the following scheme.
Chain extension polymerizations were conducted and while a slight tailing was observed but there was a clean increase in MW of the chain extended block copolymer indicating high retention of CEF during formation of the macroinitiator.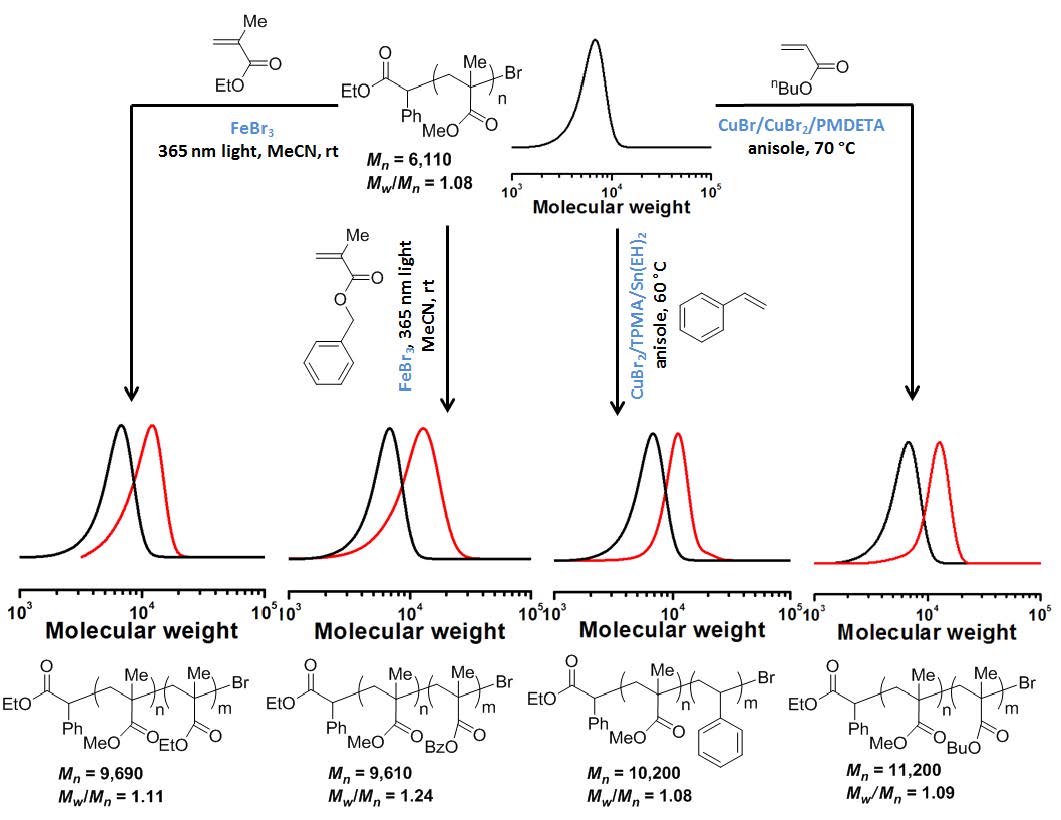
A series of runs were conducted under the following conditions: MMA:FeBr3 = X:1, MeCN 33.3%, 4.9 mW/cm2 UV hv, at room temperature with different ratios of monomer:catalyst. It was determined that a controlled polymerization occurred with ratios between 100:1 and 300:1. There was an increase in induction period and slower rate of polymerization as the concentration of FeBr3 was decreased, all systems producing polymers with low PDI.
The experiments were repeated targeting different DPs and using different monomers and the results are reported in the following table. 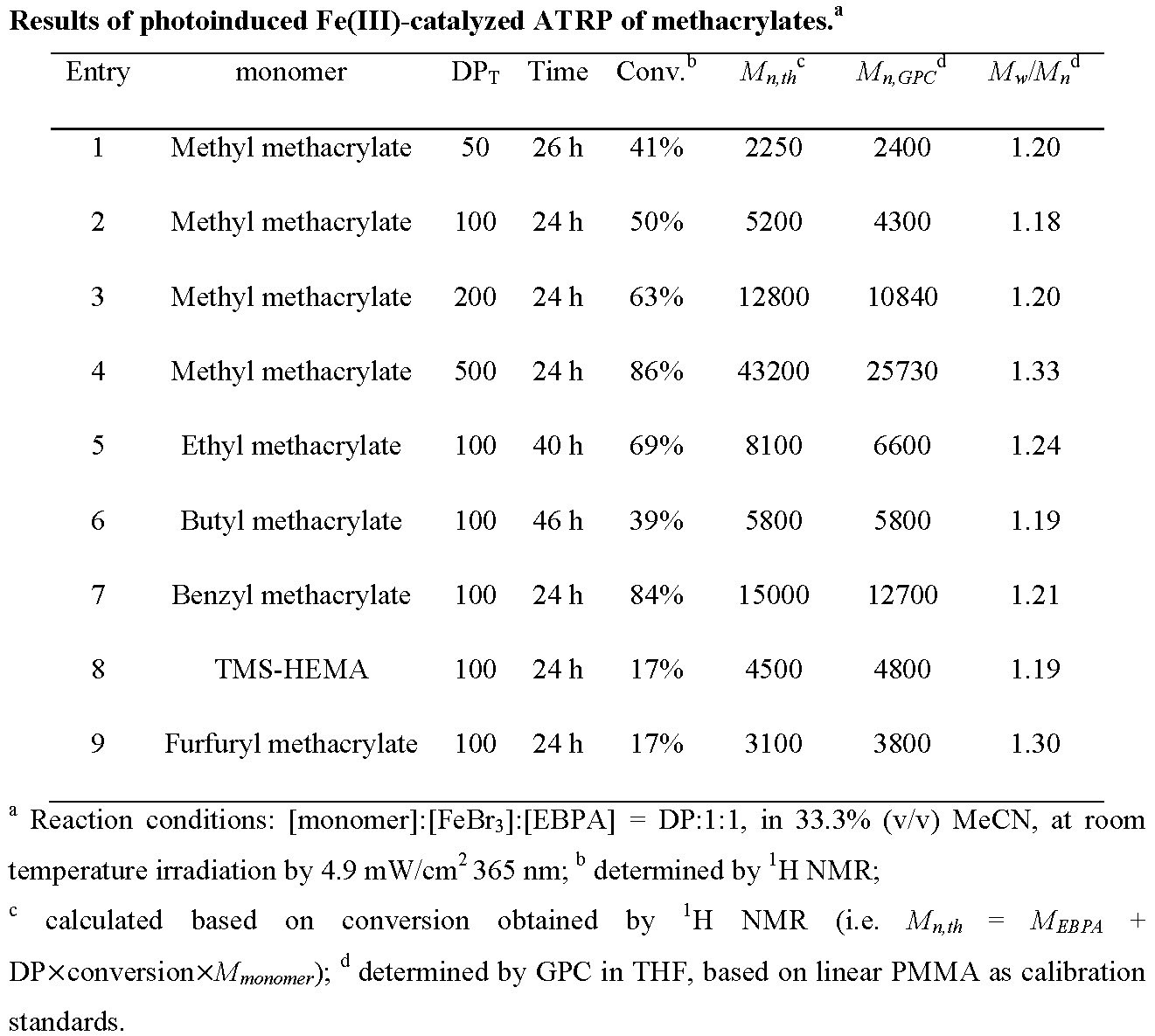
The use of an air-stable FeIII catalyst provides a facile reaction setup, and no additional ligands are required, due to the use of acetonitrile or DMF as polar solvents as they also act as solubilizing/stabilizing ligands. The low cost and availability of all reagents and simple setup make this system attractive for the preparation of polymers for some applications.
Photoinduced Metal-free ATRP
While the development of photoinduced copper mediated ATRP provided the potential for microscopic control of these photochemical reactions, due to the simple triggering by visible light and was envisioned to be an attractive feature for building useful materials with complex molecular architectures there remained a considerable interest to develop an ATRP method that does not use transition metals catalysts, especially for preparation of (co)polymers targeting electronic or biomaterials applications. Systems based on alkyl iodides and various organic catalysts were previously described (11) however, they rely on the presence of a much weaker C-I bond and the ability of iodine to form various hypervalent species. Therefore it remained a challenge to extend such metal-free catalytic systems to alkyl bromides, commonly used in ATRP.
In 2014 Hawker reported a photoinduced metal-free ATRP of methyl methacrylate using 10-phenylphenothiazine (Ph-PTZ) as an organic photocatalyst. (12) The proposed mechanism for PTZ-catalyzed photoATRP involves a three component photoredox cycle. The photoexcited PTZ* activates an alkyl halide and generates radicals, while the PTZ+•Br- specie deactivates the radical and regenerates the ground state PTZ. 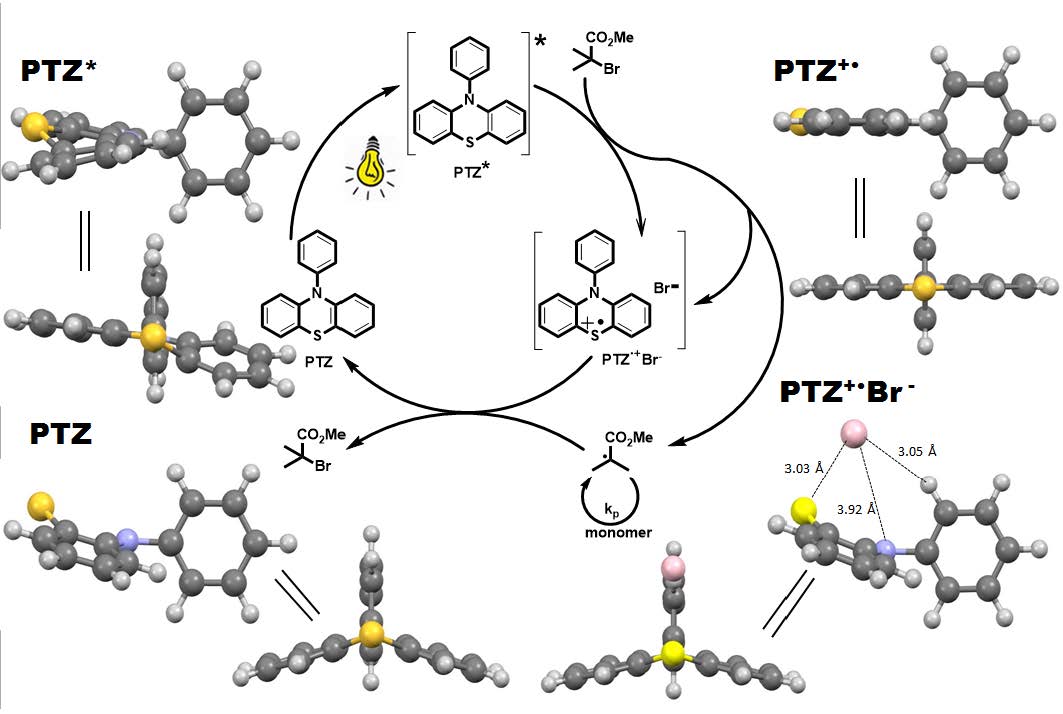
Scheme: Proposed mechanism for PTZ-catalyzed photoATRP and optimized geometries of related intermediates.
Perylene was also used as a successful photocatalyst for metal-free ATRP (13, 14) although the greater detail of the polymerization in the paper does indicate that the process displayed limited initiation efficiency, ranging from 2% to 40% in various solvents, when catalytic amounts, 0.11 equiv to initiator, of perylene were used. Although a linear semilogarithmic kinetic plot and good temporal control by light was observed, the molecular weight decreased with conversion. This could be attributed to the fact that oxidized perylene radical cation did not efficiently quench the radicals. Retention of the bromine chain end functionality was limited, which was confirmed by several chain-extension polymerizations and MALDI-TOF. This might arise because of bromine abstraction from EBPA by the propagating radical as well as other undesired radical terminations, rather than a clean deactivation pathway. The patent did claim a successful metal free photo-ATRP.
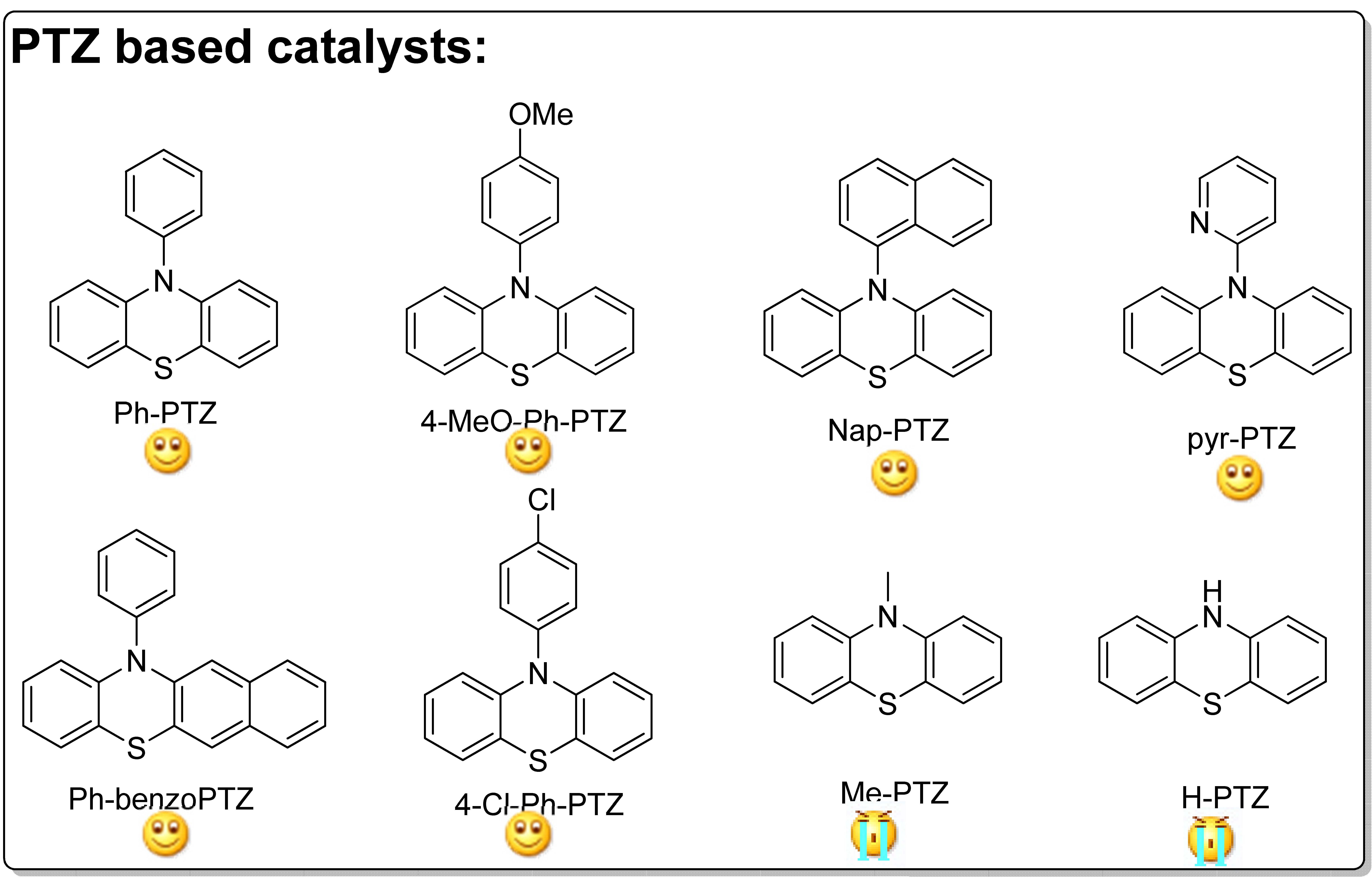
The Matyjaszewski group examined Ph-PTZ and other phenothiazine derivatives, such as 10-(4-methoxyphenyl)-phenothiazine (4-MeOPh-PTZ) and 10-(1-naphthalenyl)-phenothiazine (Nap-PTZ), as organic photocatalysts for ATRP of acrylonitrile. (15) The reason for selecting acrylonitrile as the monomer for this study was the fact that acrylonitrile based block copolymers have been recently used as precursors for N-doped nanostructured carbons that show interesting catalytic properties and excellent performance as supercapacitors, materials for CO2 capture, catalysts for oxygen reduction or counter electrodes in dye sensitized solar cells. (16-18) Since residual amounts of Cu could potentially impact the properties of the nanostructured carbons, it was of interest to evaluate properties of nanostructured carbons prepared in the absence of any transition metal catalysts.
A series of PTZ derivatives were examined for the polymerization of acrylonitrile in DMF or DMSO as solvents. The following figure shows the agents and indicates the success attained with each derivative.
CV spectra measurements indicated that when a stable radical cation was formed a controlled polymerization was observed, whereas the formation of an unstable radical cation resulted in no control over the polymerization. Measurement of the lifetime of the excited state of the radical cations varied from 4.47 ns for PTZ to 6.00 ns for 4-OMe-PTZ.
Molecular weight of the polymers followed theoretical values after 50% conversion suggesting slow but complete initiation resulting in Mw/Mn values were close to 1.5.
The following figure summarizes this paper and shows that while the Mw/Mn values were close to 1.5 chain extension of the formed PAN macroinitiator with MMA showed a clean shift in the GPC curve to higher MW providing a well-defined PAN-b-PMMA block copolymer and indicated retention of high chain end functionality in the initial PAN macroinitiator and high efficiency for cross propagation.
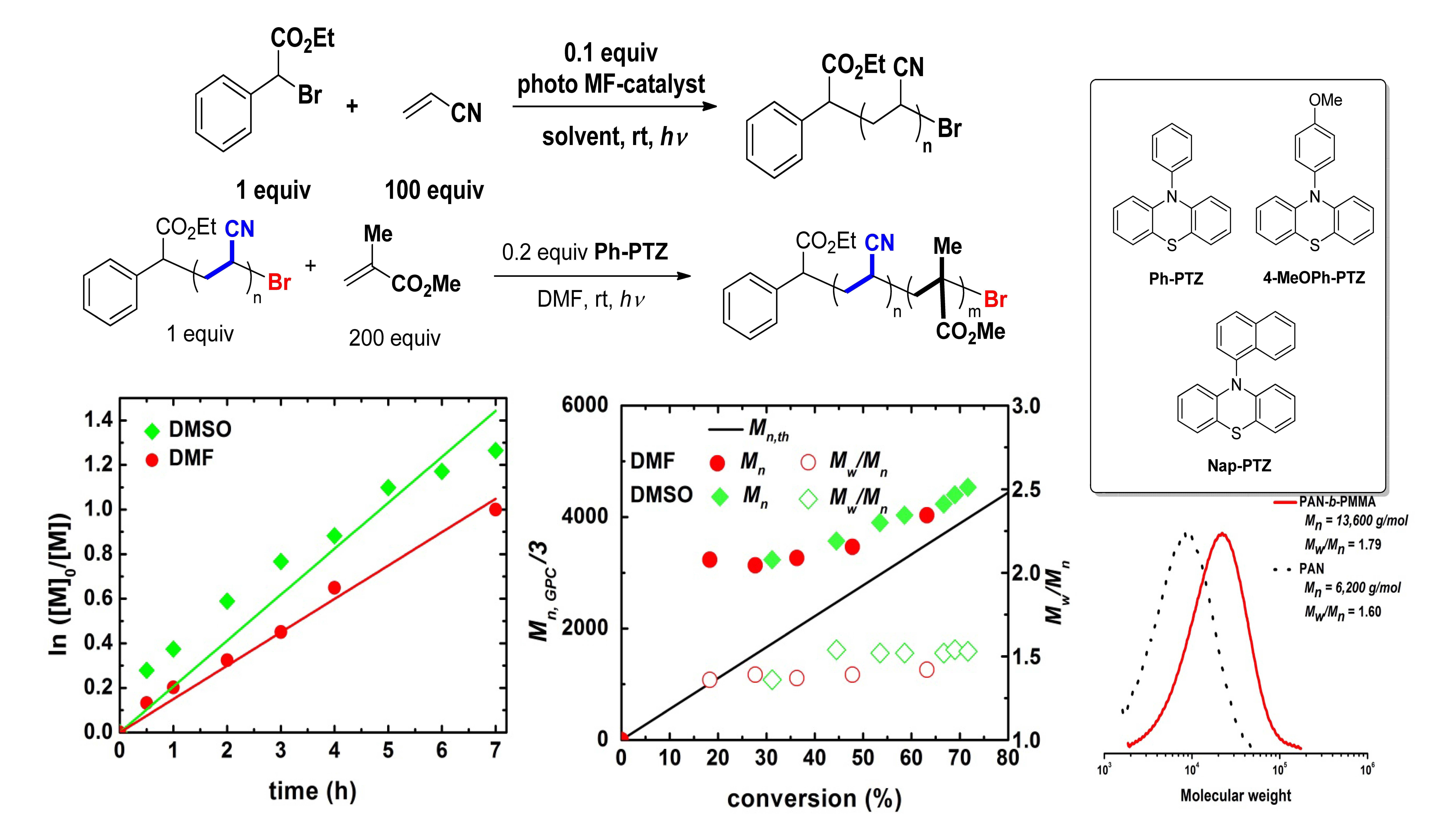
While this transition metal free ATRP has some room for improvements in initiation efficiency allowing preparation of materials with lower dispersity it currently provides materials without residual transition metal that could be useful for the synthesis of electronic materials.
An extensive investigation recently elucidated the mechanism of metal free PTZ-catalyzed photoATRP, especially in comparison to traditional Cu-mediated ATRP. (19)
The net result of extensive research directed at developing low ppm level of catalyst ATRP procedures can be summarized in the following schematic.
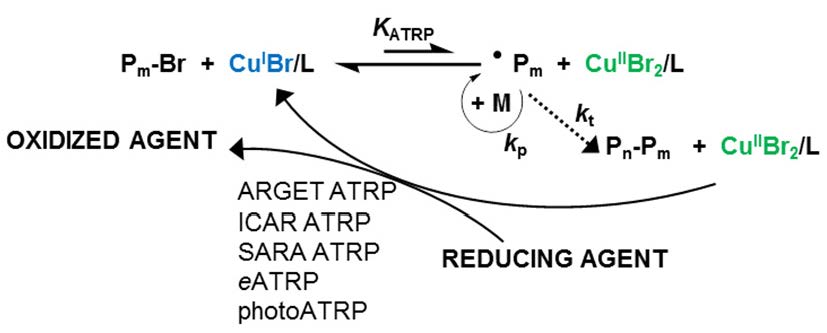
The use of low [Cu], i.e. <1% vs. initiator, and generating reaction conditions that result in a low steady state [R•], which depends on the rate of activator regeneration and termination provides a well-controlled polymerization with the additional benefit of very low loss of chain end functionality, and now includes photoATRP.
Metal-Free Surface-Initiated Atom Transfer Radical Polymerization (SI-ATRP)
A logical step after developing a photoinduced metal-free ATRP using photoexcited 10-phenylphenothiazine (PhPTZ) organic catalyst for polymerization of a wide range of monomers, including methacrylates and acrylonitrile via metal-free ATRP which eliminated residual metal in the product (19) was extension of the procedure to grafting from the surface of solid inorganic particles. Well-defined polymer-inorganic hybrid materials were prepared via metal-free surface-initiated atom transfer radical polymerization (SI-ATRP) with 10-phenylphenothiazine (PhPTZ) as the photocatalyst and 2-bromo-2-phenylacetate initiator tethered to silica surfaces. (20) The hybrid materials prepared via metal-free SI-ATRP at low catalyst concentration (0.02 or 0.1 mol %) were of quality comparable to hybrid particles prepared by Cu catalyzed SI-ATRP. TEM images confirmed successful grafting of PMMA from silica nanoparticles and show a small fraction of aggregated nanoparticles. The broad size distribution, observed in DLS, originated in broad size distribution of the pristine silica nanoparticles.
REFERENCES
(1) Pan, X.; Malhotra, N.; Simakova, A.; Wang, Z.; Konkolewicz, D.; Matyjaszewski, K. J. Am. Chem. Soc. 2015, 137, 15430-15433.
(2) Pan, X.; Malhotra, N.; Zhang, J.; Matyjaszewski, K. Macromolecules 2015, 48, 6948-6954.
(3) Tsarevsky, N. V.; Pintauer, T.; Matyjaszewski, K. Macromolecules 2004, 37, 9768-9778.
(4) Braunecker, W. A.; Tsarevsky, N. V.; Gennaro, A.; Matyjaszewski, K. Macromolecules 2009, 42, 6348-6360.
(5) Konkolewicz, D.; Magenau, A. J. D.; Averick, S. E.; Simakova, A.; He, H.; Matyjaszewski, K. Macromolecules 2012, 45, 4461-4468.
(6) Simakova, A.; Averick, S. E.; Konkolewicz, D.; Matyjaszewski, K. Macromolecules 2012, 45, 6371-6379.
(7) Konkolewicz, D.; Krys, P.; Gois, J. R.; Mendonca, P. V.; Zhong, M.; Wang, Y.; Gennaro, A.; Isse, A. A.; Fantin, M.; Matyjaszewski, K. Macromolecules 2014, 47, 560-570.
(8) Konkolewicz, D.; Schroder, K.; Buback, J.; Bernhard, S.; Matyjaszewski, K. ACS Macro Lett. 2012, 1, 1219-1223.
(9) Wang, Y.; Matyjaszewski, K. Macromolecules 2010, 43, 4003-4005.
(10) Xue, Z.; Nguyen, T. B. L.; Noh, S. K.; Lyoo, W. S. Angew. Chem., Int. Ed. 2008, 47, 6426-6429.
(11) Goto, A.; Tsujii, Y.; Fukuda, T.; Kaji, H. Pacifichem 2010, International Chemical Congress of Pacific Basin Societies, Honolulu, HI, United States, December 15-20, 2010 2010, MACRO-913.
(12) Treat, N. J.; Sprafke, H.; Kramer, J. W.; Clark, P. G.; Barton, B. E.; Read de Alaniz, J.; Fors, B. P.; Hawker, C. J. J. Am. Chem. Soc. 2014, 136, 16096-16101.
(13) Miyake, G. M.; Theriot, J. C. Macromolecules (Washington, DC, U. S.) 2014, 47, 8255-8261.
(14) Miyake, G. M. In USPTO; (California Institute of Technology, USA). Application: US 20150018446, 2015; p 18pp.
(15) Pan, X.; Lamson, M.; Yan, J.; Matyjaszewski, K. ACS Macro Lett. 2015, 4, 192-196.
(16) McGann, J. P.; Zhong, M.; Kim, E. K.; Natesakhawat, S.; Jaroniec, M.; Whitacre, J. F.; Matyjaszewski, K.; Kowalewski, T. Macromol. Chem. Phys. 2012, 213, 1078-1090.
(17) Zhong, M.; Kim, E. K.; McGann, J. P.; Chun, S.-E.; Whitacre, J. F.; Jaroniec, M.; Matyjaszewski, K.; Kowalewski, T. J. Am. Chem. Soc. 2012, 134, 14846-14857.
(18) Kim, E. K.; Zhong, M.; McGann, J. P.; Chun, S.-E.; Whitacre, J. F.; Jaroniec, M.; Matyjaszewski, K.; Kowalewski, T. Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.) 2012, 53, 227.
(19) Pan, X.; Fang, C.; Fantin, M.; Malhotra, N.; So, W. Y.; Peteanu, L. A.; Isse, A. A.; Gennaro, A.; Liu, P.; Matyjaszewski, K. J. Am. Chem. Soc. 2016, 138, 2411–2425.
(20)Yan, J.; Pan, X.; Schmitt, M.; Wang, Z.; Bockstaller, M. R.; Matyjaszewski, K. ACS Macro Lett. 2016, 5, 661-665.
Mechanically Controlled ATRP
As noted above both electricity and light have been utilized to control the activation/deactivation processes in ATRP thereby providing temporal control over the polymerization. Mechanical forces offer an opportunity to overcome the limited light absorption of the monomer or solvent as ultrasound has deeper penetration than light and is not affected by light-scattering in heterogeneous systems or gels. Interestingly, ultrasound was recently applied to mechanically controlled ATRP (mechanoATRP) in the presence of piezoelectric barium titanate nanoparticles.(1) The Esser-Khan group used “normal” ATRP ratios of reagents; i.e. 10,000 ppm of a catalyst based on Cu(OTf)2, Me6TREN and Bu4NBr in DMF with ethyl α-bromoisobutyrate as initiator for the polymerization of n-butyl acrylate, (mole ratio 1:100) under intermittent ultrasound radiation and formed polymers with low dispersity. Although high conversion was attained the molecular weight of the polymers was lower than expected (Mn < 3000) for a controlled radical polymerization.
Within our group we successfully extended a temporal controlled mechanoATRP using a low intensity ultrasound bath, in the presence of a low concentration (75 ppm) of Cu/tris(2-pyridylmethyl)amine (TPMA) catalyst in DMSO as solvent to prepare well-defined poly(methyl acrylate), PMA, with excellent control over molecular weight (up to Mn = 20,000) and dispersity (1.05 < Đ < 1.18) with various types of piezoelectric barium titanate (BaTiO3) nanoparticles, loadings of nanoparticles, and targeted degrees of polymerization. Since, ultrasound was demonstrated to disrupt metal−ligand interactions, TPMA was selected as the ligand to form stable Cu complexes. The effect of different size and shapes of BaTiO3 NPs on the polymerization were investigated, the results were summarized in the paper, and it was determined that crystal structure and particle size are critical for generating the appropriate level of piezoelectricity. The tetragonal BaTiO3 crystals are highly distorted and have larger dielectric constant ε ∼ 3000 and stronger piezoelectricity than the cubic phase BaTiO3 NPs with ε ∼ 500. Furthermore, for smaller NPs, the mechano-electric conversion can be enhanced due to the surface/interface effects and higher surface-to-volume ratios. Results showed that reactions with higher loadings of BaTiO3 NPs gave faster polymerization rate, indicating higher concentration of radicals as well as a higher ratio of CuI/CuII.
However it was also observed that the BaTiO3 NPs tend to aggregate and precipitate in the solution without ultrasound. Surface modification with poly-(methyl methacrylate) chains (Mn = 185000, graft density σ = 0.43 chain/nm2) help to stabilize the particles in solution and keep them uniformly dispersed. A polymerization using PMMA modified BaTiO3 NPs at a total loading of 4.5 wt % (2.25 wt% of BaTiO3), with respect to monomer and solvent, reached 78% conversion after 8 h, providing PMA with Mn = 13770 and Đ = 1.05.
Ultrasound stimulus was also used to demonstrate temporal control over the polymerization. This was possible due to low ppm concentration of Cu catalysts that was quickly lost the presence of the [CuI] activator due to radical termination reactions, if catalysts regeneration was stopped. The experiments with on−off ultrasound cycles demonstrated that polymerization could be stopped or restarted in response to ultrasound. A low conversion was observed in the absence of ultrasound as well as a clear continuation of the polymerization after re-exposure to ultrasound agitation. Additionally, efficient control over the polymerization was achieved as polymers with low Đ and molecular weights agreeing well with the theoretical values.
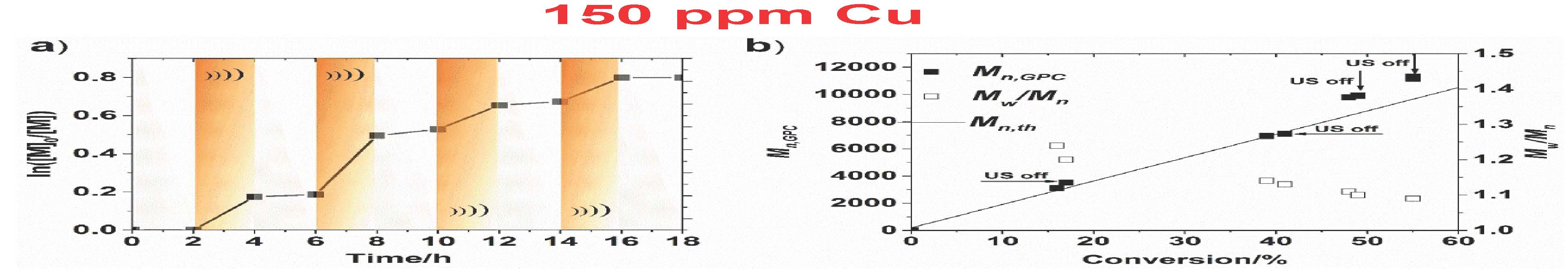
However, the reaction required a high loading of piezoelectric barium titanate particles (∼4.5 wt %) to maintain the desired polymerization rate throughout the reaction due to the inefficient electron transfer (ET) between the nanoparticles and CuII/L in the heterogeneous system. Stronger interaction between semiconductors and electron acceptors (CuII/L) can favor ET due to decreased energy of the conduction band of the semiconductor. If CuII/L is strongly bound to the piezoelectric nanoparticles, it was anticipated that mechano-induced electrons can be rapidly captured by CuII/L, resulting in CuI/L that can efficiently activate the polymerization. It was demonstrated that ET from piezoelectric nanoparticles to CuII/L is enhanced by a proper selection of the piezoelectric nanoparticles. ZnO appeared to be an excellent candidate. Indeed there was enhanced MechanoATRP using small ZnO nanoparticles since ZnO is a better piezoelectric compound. The reaction was much faster with much lower concentrations of solids in the reaction medium, 0.6% compared to 4.5%. Was this due to the better piezoelectric coefficient which is measured as D33, which quantifies the volume change when a piezoelectric material is subject to an electric field. The values are D33 (ZnO) = 3 pC/N and D33 (BaTiO3) = 10 pC/N which indicates that BaTiO3 should be more efficient. Why the discrepancy? The answer was provided by conducting a Zeta-potential analysis which indicated that there is an interaction between ZnO and CuBr2/L. This was confirmed by UV-Vis-NIR spectra which showed that the color of ZnO changed from white to brown after ultrasound and there was a faster reduction of CuII with ZnO under ultrasound.
Control experiments with ultrasound stimulation in the presence of ZnO showed a FRP occurred in the presence of ultrasound and ZnO alone however in the presence of CuII/TPMA controlled polymerizations were observed and the polymerizations were faster and better controlled in the presence of a higher concentration of ligand and the ligand was consumed in the process. Experimental molecular weights agreed well with theoretical values indicating all chains had grown from the added initiator. It was determined that there was no generation of a biradical product by cleavage of the R-X initiator under ZnO/ piezoelectric stimulation and it was also determined that CuI species did not come from the homolytic cleavage of CuII/L.
The dominant pathway is: 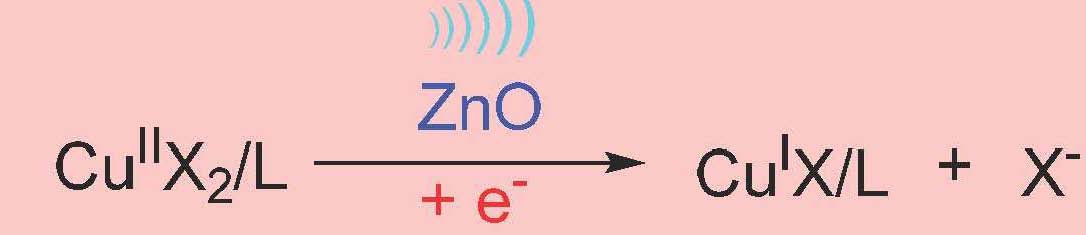
A second pathway for activation of the catalyst could be formation of radicals from cavitation of the monomer/solvent which could initiate new chains. The polymerization in the absence of CuBr2/TPMA and EBiB after 8 h ultrasonication resulted in low conversion (<15%) generating a polymer with high molecular weight (Mn > 260,000) and broad molecular weight distribution (Mw/Mn > 1.9). Therefore the actual contribution of radicals from cavitation to the mechano-ATRP is limited because polymerization reached a very low conversion (<5%) in the absence of EBiB.
A successful mechano-ATRP was carried out even at a 0.06 wt % loading of ZnO piezoelectric nanoparticles, a 75-fold decrease in the concentration of solids as compared to previous results. (3) MchanoATRP of other acrylates using ZnO was also evaluated and well controlled polymerizations of MA, EA, tBA, BA and MMA were attained. Temporal control was also demonstrated during the polymerization of MA. In addition CEF was confirmed by successful chain extension of a macroinitiator formed in a MechanoATRP. Therefore it was determined that ZnO acts as an efficient piezoelectric semiconductor. However, as with the BaTiO3 particles, the dispersion of the particles in the reaction medium is opaque due to aggregation of the dispersed particles. Efficiencies of bare ZnO and ZnO with solubilizing octylamine (OA) and grafted poly(methyl methacrylate) (PMMA) ligands were compared. Polymerizations were examined with OA-ZnO (5 nm) dispersion, which is transparent indicating the solubility of the hybrid particle in the polymerization medium.
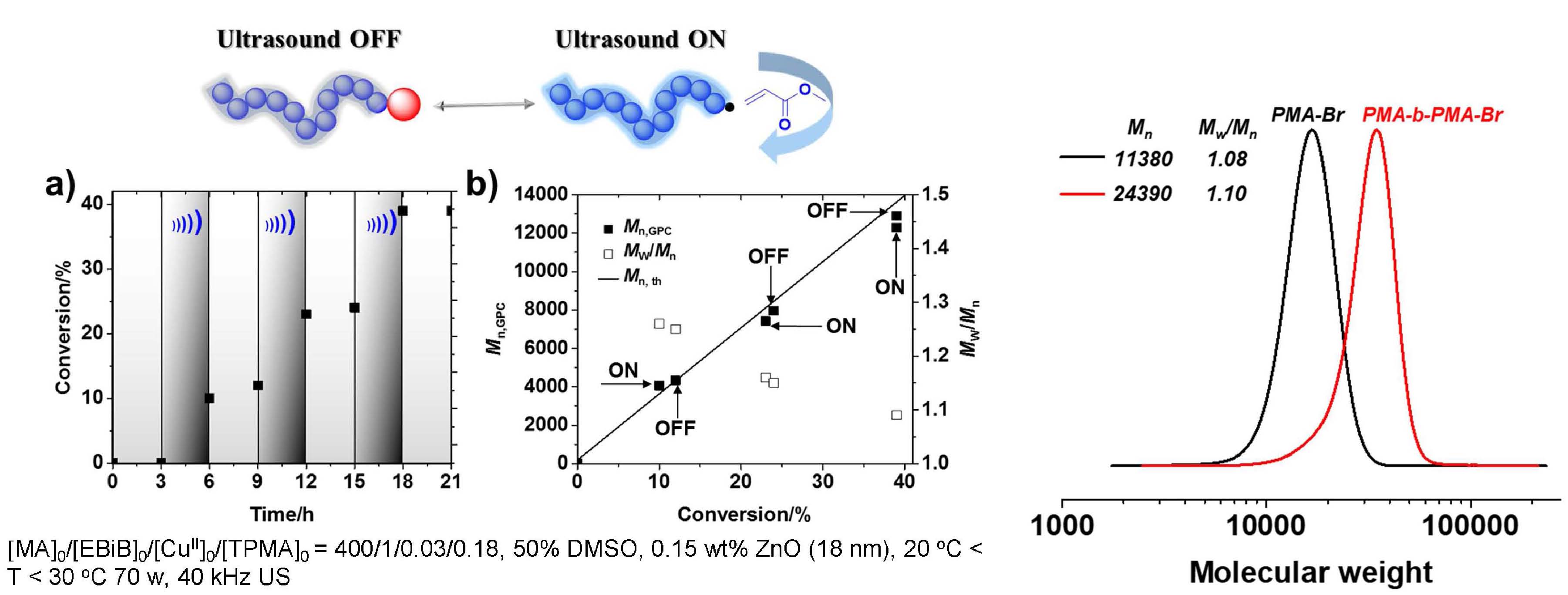
PhotoATRP with OA-ZnO (5 nm) under UV light in different solvents showed that the reaction was faster in DMSO than anisole but both solvents resulted in well controlled polymerizations. The polymerization in DMSO was also successful under violet light and was faster in the presence of the OA-ZnO nanoparticles. MechanoATRP with OA-ZnO (5 nm) also produced well controlled polymerizations MechanoATRP with OA-ZnO (5 nm) in the presence of different concentrations of OA-ZnO also were well controlled with the rate dependent of the concentration of OA-ZnO. In summary low ppm of Cu catalyst allows temporal control over polymerization in ultrasound mediated ATRP. Ligand stabilized particles are more efficient and the interaction between ZnO and CuIIX2/L favored the electron transfer induced by ultrasound. The polymerization with 5 nm OA-ZnO could be controlled by light and ultrasound.
Since the above reactions were heterogeneous and carried out in organic media, it was desirable to extend the procedure and develop homogeneous ultrasound-induced aqueous ATRP (Sono-ATRP), which would provide a new platform to synthesize water-soluble polymers for biomedical applications under mild conditions.
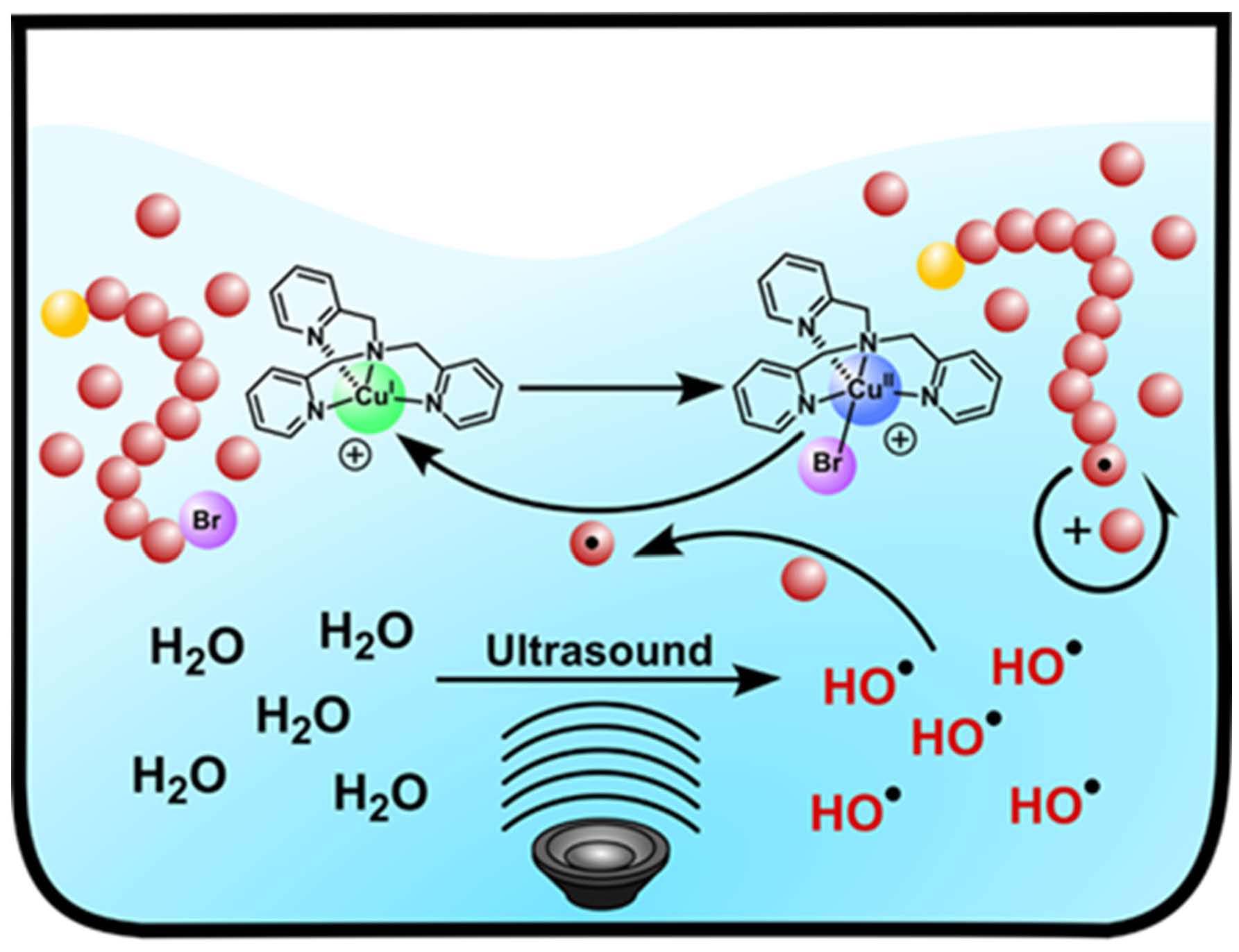
The key requirement for establishing an aqueous sono-ATRP is the continuous regeneration of the CuI species during the ultrasonication. When an ultrasonic wave propagates through aqueous media, hydroxyl radicals are produced because of the acoustic cavitation which provides the primary mechanism for sonochemical effects. These radicals can react with a vinyl monomer to form carbon radicals and initiate a radical polymerization. Using this strategy, we used an ultrasound probe to in situ initiate the emulsion free radical polymerization of n-butyl acrylate without addition of any conventional initiators. (4) The paper reported the extension of ultrasound-mediated ATRP to aqueous media using ppm level of Cu/tris(2-pyridylmethyl)amine (TPMA) catalyst, without any additional conventional initiators or reducing agents. Well defined polymers with high molecular weight (Mn > 240,000), low dispersity, and high chain-end functionality were prepared. This aqueous sono-ATRP utilizes ppm level catalyst in aqueous media under benign conditions, i.e., using simple agitation in an ultrasonic bath at room temperature. Oligo(ethylene oxide) methyl ether methacrylate, Mn = 500 (OEOMA500), was polymerized in water under sonication (40 kHz, 110 W) using poly(ethylene oxide)−bromophenylacetate (PEO2k-BPA) as a macroinitiator. CuIIBr2/TPMA was selected as the catalyst since it forms a stable complex without any significant disproportionation in aqueous media. The addition of sodium bromide (NaBr) to the solution minimized the dissociation of bromide anions from the deactivators. An ultrasonic bath was used to activate the polymerization because of easy scalability and low intensity when targeting high molecular weight. Without NaBr, the reaction provided a poorly controlled polymerization; i.e., molecular weight decreased with increasing conversion. This can be attributed to the dissociation of the halide anion from the CuBr2/TPMA deactivator, lowering the concentration of deactivators. The presence of excess NaBr maintained a sufficient concentration of deactivators, providing a better control over the polymerization. The experimental Mn values were slightly lower than the theoretical value in the aqueous sono-ATRP of OEOMA500. Plausibly, hydroxyl radicals and not only continuously reduced CuII species but also reacted with monomer and generated some new polymer chains throughout the reaction.
Ethanol can react with hydroxyl radicals to produce carbon based radicals. Thus, after addition of ethanol, the CuII species could be more efficiently reduced by these radicals. Additionally, the introduction of ethanol reduces the water concentration, yielding a lower concentration of hydroxyl radicals. The polymerization performed in ethanol/water showed a better agreement between theoretical and experimental molecular weights than that conducted in water. A polymerization with 400 ppm of Cu in 1/1 ethanol/water showed a linear semilogarithmic kinetic plot, affording a well-defined polymer with Mn = 83,960 and Đ = 1.19 after 4 h ultrasonication.
This procedure was also extended to the synthesis of bioconjugates using a DNA-based macroinitiator prepared according to the previous work. (5) The DNA oligonucleotide (SeqAA, 5′-ATC TGA GAC TCA CTG-3′, Mw = 5365 based on MALDI) was prepared with a Cy3 dye at the 3′-end to facilitate the visualization of the hybrid and to confirm that the DNA strand was not cleaved under ultrasonication. The 5′-end included an α-bromoisobutyrate (iBBr) group to initiate the polymerization. The synthesis of the DNA−polymer biohybrid was carried out under highly diluted conditions. OEOMA500 was used as the monomer to perform the polymerization in aqueous media. SeqAA-b-POEOMA, bioconjugate was prepared in a well-controlled manner, providing a polymer with Mn = 36 250 and with a dispersity Mw/Mn = 1.41. GPC traces clearly showed that the molecular weight of the DNA−polymer hybrid shifted to a higher range from the original DNA macroinitiator.
In summary, a successful ultrasonication-induced aqueous ATRP with ppm level Cu catalyst in an ultrasonic bath enabled polymerization with excellent control, providing polymers with high molecular weight and low dispersity. The low intensity ultrasonic bath was used to continuously regenerate activators and also to prevent chain rupture by shear forces,18 allowing the synthesis of high molecular weight water-soluble polymers.
1. Mohapatra, H.; Kleiman, M.; Esser-Kahn, A. P., Mechanically controlled radical polymerization initiated by ultrasound. Nature Chemistry 2017, 9, 175.
2. Wang, Z.; Pan, X.; Yan, J.; Dadashi-Silab, S.; Xie, G.; Zhang, J.; Wang, Z.; Xia, H.; Matyjaszewski, K., Temporal Control in Mechanically Controlled Atom Transfer Radical Polymerization Using Low ppm of Cu Catalyst. ACS Macro Lett. 2017, 6 (5), 546-549.
3. Wang, Z.; Pan, X.; Li, L.; Fantin, M.; Yan, J.; Wang, Z.; Wang, Z.; Xia, H.; Matyjaszewski, K., Enhancing Mechanically Induced ATRP by Promoting Interfacial Electron Transfer from Piezoelectric Nanoparticles to Cu Catalysts. Macromolecules 2017, 50, 7940.
4. Wang, Z.; Wang, Z.; Pan, X.; Fu, L.; Lathwal, S.; Olszewski, M.; Yan, J.; Enciso, A. E.; Wang, Z.; Xia, H.; Matyjaszewski, K., Ultrasonication-Induced Aqueous Atom Transfer Radical Polymerization. ACS Macro Lett. 2018, 7, 275-280.
5. Averick, S. E.; Dey, S. K.; Grahacharya, D.; Matyjaszewski, K.; Das, S. R. Solid-Phase Incorporation of an ATRP Initiator for Polymer−DNA Biohybrids. Angew. Chem., Int. Ed. 2014, 53 (10), 2739−2744.