Networks/Gels
Functional networks:
Physically crosslinked gels:
In this section we will extend the discussion on the concurrent copolymerization of a monomer and a divinyl cross-linker using ATRP to generate branched copolymers and/or networks/gels. The experimental gel points based on conversion of the monomer and cross-linker are determined and compared to each other. In the cited papers the dependence of experimental gel points on various parameters, including the molar ratios of cross-linker to initiator, the concentrations of reagents, the structures of monomer and cross-linker, and the reactivity of the vinyl groups present in the cross-linker and monomer, were systematically studied.(1-4)
This work detailed below is, similar to the section on star copolymers, predominately based on the work of Haifeng Gao reported in his 2008 PhD thesis and in a series of papers based on the studies. His studies on the topology of copolymer networks prepared in a single-batch concurrent copolymerization of a monomer and a cross-linker can be viewed as an extension of the sequential copolymerization reactions discussed above for arm first and core first strategies for synthesis of star copolymers. In this section we provide further discussion on how such controlled copolymerization reactions can be utilized to form networks with topologies depending on when the crosslinker is added to the copolymerization and the reactivity of each vinyl group in the crosslinker.
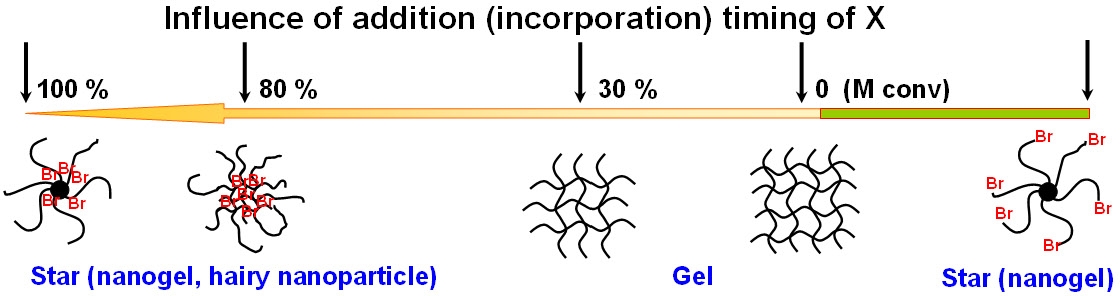
Scheme A: Influence of the sequence of addition and incorporation of cross-linker (X) and monomer on polymer architecture during the ATRcoP of monomer (M) and cross-linker using R-Br as initiator
During the radical copolymerization of a divinyl cross-linker, the cross-linkage, provided by a cross-linker with both vinyl groups reacted, is formed in the polymer chains via reaction of pendant vinyl groups with propagating chain-end radicals either inter-molecularly or intra-molecularly. In the presence of an appropriate mole fraction of crosslinker the molecular weight and/or size of the branched polymers increase exponentially with the progress of inter-molecular cross-linking reactions, and finally reach an "infinite" value with the formation of a polymeric network (gel). The transition from viscous liquid containing hyper-branched macromolecules, a sol, to an elastic gel is defined as the "gel point".
The properties of gels prepared by standard free radical polymerization and controlled radical copolymerization procedures differ. Highly branched polymers and/or gels with inhomogeneous structures are formed during most conventional RP reactions because of the intrinsic features of the RP method, including slow initiation, fast chain propagation, and exclusive radical termination reactions. A consequence of the extremely low polymer concentration at the beginning of the copolymerization in a standard FRP is that polymer chains seldom overlap and most of the pendant vinyl groups are consumed via intramolecular cyclization, ultimately producing highly cross-linked nanogels, see the upper schematic in the following Scheme B. As the reaction proceeds, the number of these nanogels increases and consequently, the radicals generated later in the reaction connect these preformed overlapped nanogels into large molecules and form a heterogeneous gel network.
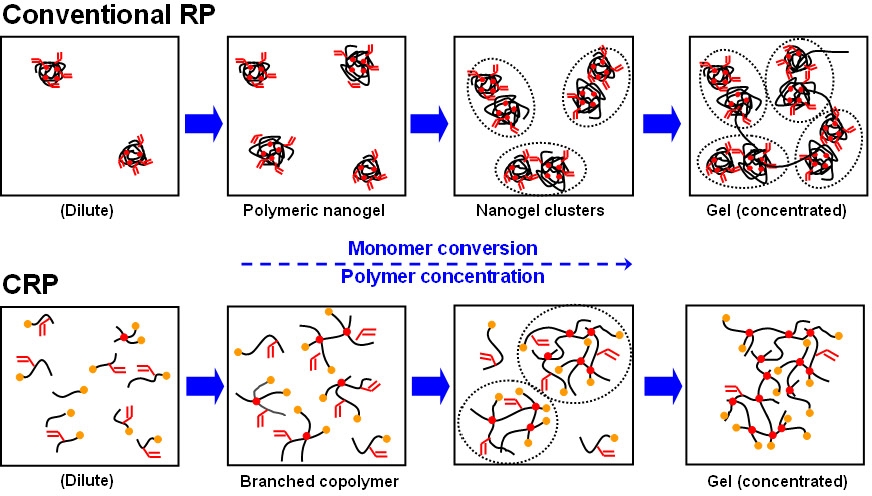
Scheme B: Differeces in the gelation process between RP and CRP
In contrast to the conventional RP technique, the CRP techniques possess several advantages when targeting the preparation of more homogeneous polymer networks, due to their fast initiation and rapid reversible deactivation reactions. Fast initiation, relative to propagation reactions, result in rapid conversion of all initiators into primary chains and a nearly constant number of growing primary chains throughout the polymerization, Scheme C. Therefore, the concentration of primary chains (PC) in a well controlled CRP system is similar to the concentration of added initiators throughout the polymerization (e.g., [PC]t ~ [Initiator]0 ~ mM).(5)
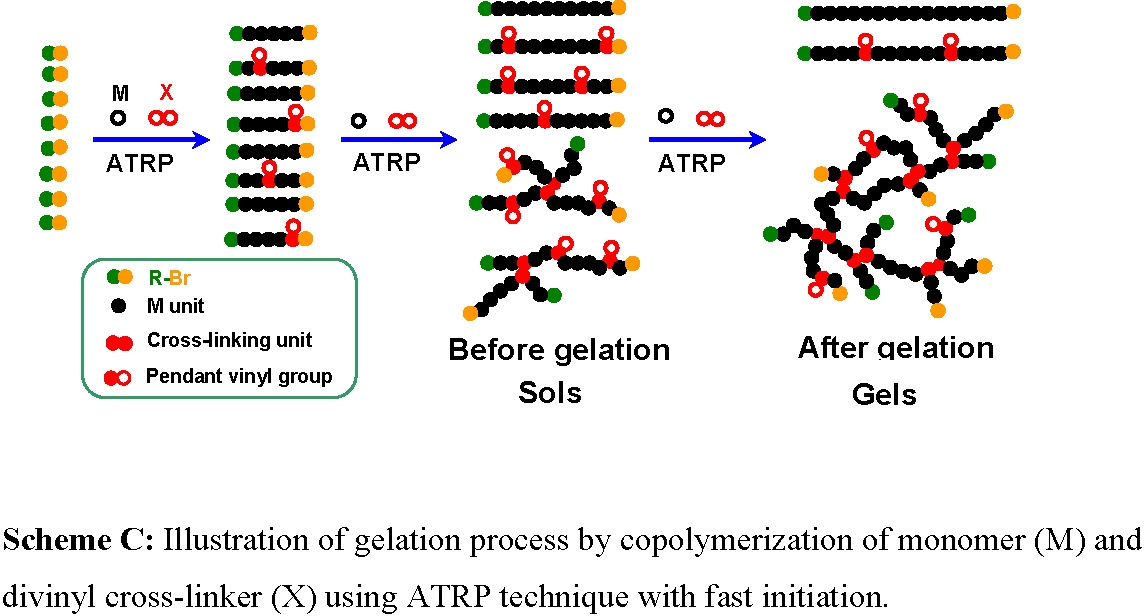
Conditions should be selected to establish an appropriate equilibrium between the rate of activation and deactivation to ensure that only a few monomer units are incorporated into the polymer chains in each activation/deactivation cycle. The resulting relatively long "dormant" period, during which time the polymer chains cannot propagate, ensures that the chains can diffuse and relax. Therefore the probability of reaction of each vinyl species: monomer, cross-linker or pendant vinyl group, is statistically determined by their concentrations in the reaction medium. Thus, the application of CRP techniques to copolymerization of monomer and cross-linker should result in a more homogeneous incorporation of branching points into the soluble (hyper)branched copolymers and a much more regular network structure in the insoluble gels, compared to polymers synthesized by conventional RP methods from similar concentrations of comonomers.(6)
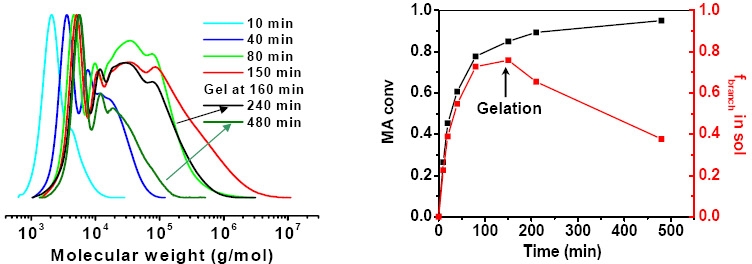
This work can be considered to be an extension of the observations of Sherrington(7) and Armes(8) who noted that if an excess of crosslinker vs. initiator is used in an ATRP, then a crosslinked network or gel will be formed if the structure of each vinyl group present in the crosslinker and monomer are similar; i.e. reactivity ratio's are close to 1 and conversion is driven to a point where the concentration of reacted pendant vinyl groups is larger than the number of primary chains. However they claimed that all linear chains disappeared at the later branching stage in the gelation process. Our results differ and indicate that during the chain propagation and branching process, linear primary chains always coexist with branched polymers in the sols and, even after gelation, linear polymer chains and branched macromolecules were still detectable in the sols by GPC measurement.
A series of cross-linked polymers were synthesized by copolymerization of different acrylate monomers and di(meth)acrylate crosslinkers including ethylene glycol diacrylate (EGDA), ethylene glycol dimethacrylate (EGDMA), and an asymetrical cross-linker (EGAMA) containing one acrylate group and one methacrylate group.
The variation of experimental gel points with different monomers and/or cross-linker conversions was systematically studied in an all acrylate system by changing three parameters: monomer concentration, structure of cross-linkers, and structure of monomers. The gel points based on conversions and the gel structural compactness did not change significantly by varying the monomer and/or cross-linker species within the scope of our study.(1) The methacrylate groups present in EGDMA crosslinker would have higher reactivity than the acrylate groups in MA and EGDA which leads to faster incorporation of the cross-linker unit into the copolymer and rapid generation of branching points, an acceleration effect. However, the resulting branched polymers, containing a gradient distribution of pendant vinyl groups and branching points resembled a starlike structure, in which the pendant vinyl groups, were located in the more densely cross-linked core. Consequently they were isolated by the surrounded shell which reduced the probability of the intermolecular reactions and retarded gelation, a deceleration effect.
These two effects intercorrelated with each other and had a significant affect on the experimental gel points of the poly(MA-co-EGDMA) system, depending on the amount of added EGDMA. When more EGDMA was added, the acceleration effect became dominant, leading to earlier gelation, at lower MA conversion, compared to the ATRcP by using the same molar amount of EGDA. These two effects became much weaker when the asymetric cross-linker EGAMA was used, because the majority of the pendant vinyl groups were acrylate groups, which had a similar reactivity to free MA monomer and slower branching compared to pendant methacrylate groups.(9)
Three initiators with progressively increased initiation efficiency, Et 2-bromopropionate (EBrP) < Et 2-bromoisobutyrate (EBiB) < 2-bromopropionitrile (BPN), were used for the ATRcP of MMA and ethylene glycol dimethacrylate (EGDMA). Due to the low initiation efficiency of EBrP gelation occurred when the molar ratio of EGDMA cross-linker to EBrP was as low as 0.25. In contrast, no gelation was observed when the most efficient initiator, BPN, was used even when the molar ratio of cross-linker to initiator was equal to unity. The use of a poorer solvent for copper catalyst also decreased the initiation efficiency and resulted in a gelation at lower monomer conversion.
The dependence of the observed gel points on the polydispersity of the primary chains was studied by using activators regenerated by electron transfer (ARGET) ATRP for copolymerization of methyl acrylate and ethylene glycol diacrylate (EGDA). Decreasing the copper concentration from tens of ppm to a few ppm broadened the polydispersity of primary chains, which resulted in an earlier gelation at lower monomer conversion.(2)
When a constant molar ratio of reagents was copolymerized under more dilute conditions there was a significant increase the fraction of intramolecular cyclization reactions which delayed the occurrence of macroscopic gelation.
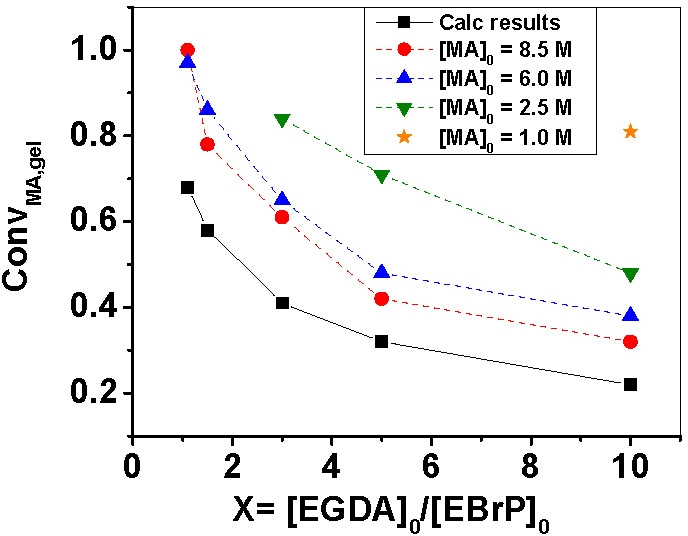
Zhu also studied the homopolymerization of ethylene glycol dimethacrylate (EGDMA) and copolymerization of methyl methacrylate (MMA) and dimethacrylate cross-linker using bulk ATRP(10,11) and found that most of the pendant vinyl groups were consumed and the cross-link density of the final gel network was close to the maximum values achievable for the added amounts of cross-linker. These results suggested that the occurrence of gelation (the gel point) in an ATRP was determined by the initial molar ratio of cross-linker to initiator. When the concentration of initially added cross-linker was larger than that of initiator, insoluble gels were produced at high (co)monomer conversions.(12) A more recent paper compared the reaction kinetics and gelation behaviors for copolymerization of oligo(ethylene glycol) methyl ether methacrylate and oligo(ethylene glycol) dimethacrylate in atom transfer, reversible addition-fragmentation chain transfer and conventional free radical copolymerization(13) and, as noted above, there was a significant difference between the gelation behavior of the FRP reaction and the CRP reactions. A significant amount of gel materials was collected before the onset of macro-gelation, detected by an abrupt increase in complex viscosity, in the FRP system whereas, in RAFT and ATRP, no gels were found until the systems approached their macro-gelation points.
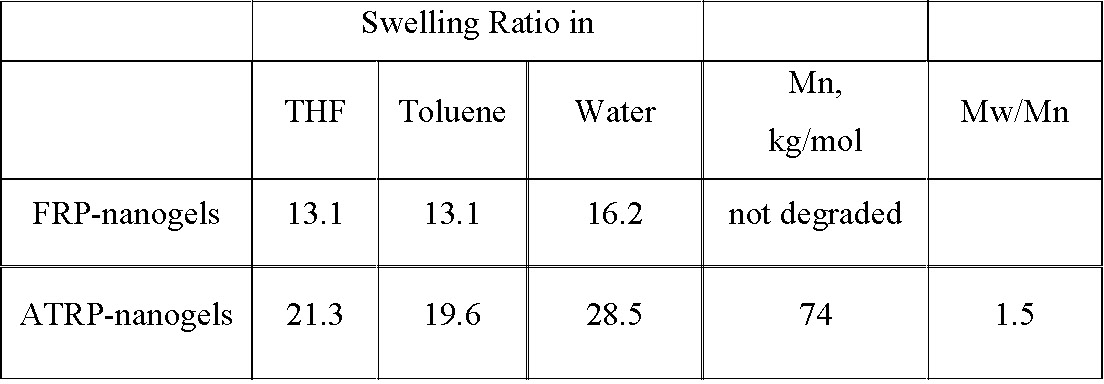
The gel materials thus synthesized by ATRP and RAFT are expected to be more homogeneous in network structure than that by FRP. This has been confirmed when the swelling behavior of a gel prepared by FRP is compared to a gel prepared by ATRP. The increased inhomogeneity of the gels prepared by FRP were confirmed when crosslinkers containing a degradable unit between the vinyl units were used to prepare gels by FRP and ATRP. The later were degradable while the gels prepared by FRP were not degraded.(14)
The "arm first" approach to stars can also be adapted to the formation of well defined copolymer networks. This was accomplished by using a difunctional initiator in an ATRP to form telefunctional polymers followed by linking of the end groups by addition of a divinyl crosslinker.(15) The amount of soluble polymer decreased with continued reaction time after addition of the crosslinker and the swelling ratio of the formed gel decreased until it reached a plateau value as the dispersed crosslinked nodes formed. However, the cross-linking density was often far below that of the theoretical value.
Presently unexamined variables include:
(a) the concentration of the polymeric precursor,
(b) the molar mass of the polymeric precursor, and
(c) the molar concentration ratio [cross-linking agent]/[polymer precursor].
This approach to the formation of well defined networks was also adapted to formation of gels with controlled topology using "click" chemistry.(16) ATRP was conducted from a bifunctional initiator then the bromine end groups of the resulting telechelic polymer were converted to azides, and crosslinking of this telechelic azido-macromonomer with a multi-acetylene functionalized small molecule via copper-catalyzed azide-alkyne cycloaddition was employed to prepare model networks. Model networks derived from a trifunctional alkyne were found to be more completely cross-linked than those derived from a tetrafunctional alkyne, presumably due to less steric hindrance in the former system.
Functional networks:
When functional groups are incorporated into the crosslinker functional gels are prepared. A degradable disulfide crosslinker was used to prepare a degradable gel by the copolymerization of MMA and a disulfide-containing difunctional methacrylate, bis(2-methacryloyloxyethyl) disulfide.(17) The gels were reduced with tributylphosphine to form soluble low molecular weight linear poly(methyl methacrylate) fragments containing thiol groups at the chain end and along the backbone, originating from the disulfide difunctional initiator and monomer. The disulfide cross-linked gels were further used as \"supermacroinitiators\" for the bulk ATRP of styrene at 90 0C, forming gels with core/shell structure that swelled more than the starting polymethacrylate gels in both THF and toluene. When this synthetic strategy is employed to copolymerize water soluble monomers in an inverse miniemulsion ATRP functional nano-sized colloidal particles are formed.(14) Dynamic light scattering (DLS) and atomic force microscopy (AFM) measurements indicated that these particles possessed excellent colloidal stability.
ATRP in inverse miniemulsion led to materials with several desirable features. The colloidal particles preserved a high degree of halogen chain-end functionality, which enabled further functionalization. Cross-linked nanogels with a uniformly cross-linked network were prepared that were degraded to individual polymeric chains with relatively narrow Mw/Mn < 1.5 in a reducing environment. Higher colloidal stability, higher swelling ratios, and better controlled degradability indicated that the nanogels prepared by ATRP were superior to their corresponding counterparts prepared by conventional FRP in inverse miniemulsion. The colloids were examined for drug delivery as detailed in page 11.
A further extension of the topology of polymer networks involved the use of inimers and multivinyl cross-linkers as branching reagents for in atom transfer radical (co)polymerizations with methyl acrylate. A fully reacted 2-((2-bromopropionyl)oxy)ethyl acrylate (BPEA) inimer produces a "T" shaped cross-linkage, from which three chains emanate. In contrast, the reactions of a divinyl cross-linker, ethylene glycol diacrylate (EGDA) and trivinyl cross-linker, 1,1,1,-trimethylolpropane triacrylate (TMPTA) generated "X" shaped and "Ж" shaped cross-linkages, respectively, from which 4 and 6 chains radiated out. The ATRP of methyl acrylate and BPEA produced soluble branched copolymers, but the addition of a small amount of EGDA into the reaction induced gelation and produced gels containing both "T" shaped and "X" shaped cross-linkages ("T&X" reaction). Three types of ATRP reactions, "T&X", "X" and "Ж" reactions, were performed to study the effect of cross-linkage structure on the experimental gel points and the properties of gels. Under the same molar ratio of [vinylbranch]0/[R-Br]0, the "T&X" reactions, experimental gelation occurred at lower monomer conversions in the "T&X" and "Ж" reactions than in the "X" reactions due to the higher intermolecular because of the faster intermolecular reactions in the "T&X" reactions. The earlier gelation in the "Ж" reaction, with respect to a comparable "X" reaction, was ascribed to the higher crosslinking efficiency and lower amount of cross-linker required for gelation when using a trivinyl cross-linker.
Consequently, due to the difference in the functionality of branching reagents, the gels obtained at complete conversion in these comparable reactions displayed a sequence of gel fraction as "T&X" ~ "Ж" > "X" and of GSR as "T&X" > "X"> "Ж", due to the difference in the structure of branching reagents.(4)
Physically crosslinked gels:
In addition to chemically crosslinked networks physically crosslinked gels were also prepared. Indeed the first gels prepared by a CRP/FRP were physically crosslinked copolymers with a water soluble backbone prepared by standard FRP grafting through well-defined vinyl macromonomers of polystyrene prepared using vinyl chloroacetate as an initiator in an ATRP. Since styrene and vinyl chloroacetate do not copolymerize, no branching or incorporation of the initiator into the backbone was observed. The macro-monomers used were of sufficiently high molecular weight to form physical crosslinks in solvents which favor the hydrophilic NVP, such as water, which prevent the copolymer from dissolving and cause it to swell. These materials, therefore, formed hydrogels of swell-abilities in water exceeding 95%, depending on the amount of styrene that was incorporated into the copolymer.(18)
Targeted applications for degradable networks or hydrogels include tissue engineering scaffolds,(19,20) responsive polymers in controlled drug delivery,(21,22) biocompatible wound dressings,(23) and coatings that impart antifouling properties.(24)
REFERENCES
(1) Gao, H.; Li, W.; Matyjaszewski, K. Macromolecules 2008, 41, 2335-2340.
(2) Li, W.; Gao, H.; Matyjaszewski, K. Macromolecules 2009, 42, 927-932.
(3) Gao, H.; Polanowski, P.; Matyjaszewski, K. Macromolecules 2009, 42, 5925-5932.
(4) Gao, H.; Min, K.; Matyjaszewski, K. Macromolecules, ACS ASAP.
(5) Braunecker, W. A.; Matyjaszewski, K. Progress in Polymer Science 2007, 32, 93-146.
(6) Ide, N.; Fukuda, T. Macromolecules 1999, 32, 95-99.
(7) Bouhier, M.-H.; Cormack, P. A. G.; Graham, S.; Sherrington, D. C. Journal of Polymer Science, Part A: Polymer Chemistry 2007, 45, 2375-2386.
(8) Bannister, I.; Billingham, N. C.; Armes, S. P.; Rannard, S. P.; Findlay, P. Macromolecules 2006, 39, 7483-7492.
(9) Gao, H.; Miasnikova, A.; Matyjaszewski, K. Macromolecules 2008, 41, 7843-7849.
(10) Wang, A. R.; Zhu, S. Polymer Engineering and Science 2005, 45, 720-727.
(11) Yu, Q.; Zhang, J.; Cheng, M.; Zhu, S. Macromolecular Chemistry and Physics 2006, 207, 287-294.
(12) Qin, Z.-q.; Zhou, M.; Sun, Y.; Yu, Q. Jiangsu Gongye Xueyuan Xuebao 2007, 19, 26-29.
(13) Yu, Q.; Xu, S.; Zhang, H.; Ding, Y.; Zhu, S. Polymer 2009, 50, 3488-3494.
(14) Oh, J. K.; Tang, C.; Gao, H.; Tsarevsky, N. V.; Matyjaszewski, K. Journal of the American Chemical Society 2006, 128, 5578-5584.
(15) Asgarzadeh, F.; Ourdouillie, P.; Beyou, E.; Chaumont, P. Macromolecules 1999, 32, 6996-7002.
(16) Johnson, J. A.; Lewis, D. R.; Diaz, D. D.; Finn, M. G.; Koberstein, J. T.; Turro, N. J. Journal of the American Chemical Society 2006, 128, 6564-6565.
(17) Tsarevsky, N. V.; Matyjaszewski, K. Macromolecules 2005, 38, 3087-3092.
(18) Matyjaszewski, K.; Beers, K. L.; Kern, A.; Gaynor, S. G. J. Polym. Sci., Part A: Polym. Chem. 1998, 36, 823-830.
(19) Siegwart, D. J.; Bencherif, S. A.; Srinivasan, A.; Hollinger, J. O.; Matyjaszewski, K. Journal of Biomedical Materials Research, Part A 2008, 87A, 345-358.
(20) Atzet, S.; Curtin, S.; Trinh, P.; Bryant, S.; Ratner, B. Biomacromolecules 2008, 9, 3370-3377.
(21) Oh, J. K.; Drumright, R.; Siegwart, D. J.; Matyjaszewski, K. Progress in Polymer Science 2008, 33, 448-477.
(22) Bajpai, A. K.; Shukla, S. K.; Bhanu, S.; Kankane, S. Progress in Polymer Science 2008, 33, 1088-1118.
(23) Madsen, J.; Armes, S. P.; Bertal, K.; Lomas, H.; MacNeil, S.; Lewis, A. L. Biomacromolecules 2008, 9, 2265-2275.
(24) Bozukova, D.; Pagnoulle, C.; De Pauw-Gillet, M.-C.; Ruth, N.; Jerome, R.; Jerome, C. Langmuir 2008, 24, 6649-6658.