Graft Copolymers
Grafting through
Grafting from
Grafting to
Once again we will only discuss a few of many graft copolymers that have been, or could be prepared by applying ATRP to materials synthesis. An early review on the preparation of graft copolymers by ATRP utilizing both "grafting through" and "grafting from" provided examples of poly(N-vinlpyrrolidone-g-styrene); poly(methyl methacrylate-g-dimethylsiloxane), discussed in greater detail below; poly(methyl methacrylate-g-lactide); poly(n-butyl acrylate-g-methyl methacrylate); and poly(n-butyl acrylate-g-ethylene) prepared by grafting through, and examples prepared by grafting from functionalized poly(isobutene), polyethylene, poly(vinyl chloride), poly(vinylidene fluoride).(1,2)
With the development of various "click" chemistry reactions graft copolymers have also been prepared by "grafting to".
Grafting through:
In a "grafting through" copolymerization the reactivity ratio of monomers and macromonomers may be affected by micro-inhomogeneity of the reaction mixture in addition to the reaction mechanism. This has often been observed in macromonomer copolymerization, or "grafting through" copolymerization for preparation of graft copolymers that would be expected to undergo phase separation. For example, when MMA was copolymerized with a poly(dimethylsiloxane)-methacrylate macromonomer, the composition of the product was strongly affected by the reaction conditions.(3)
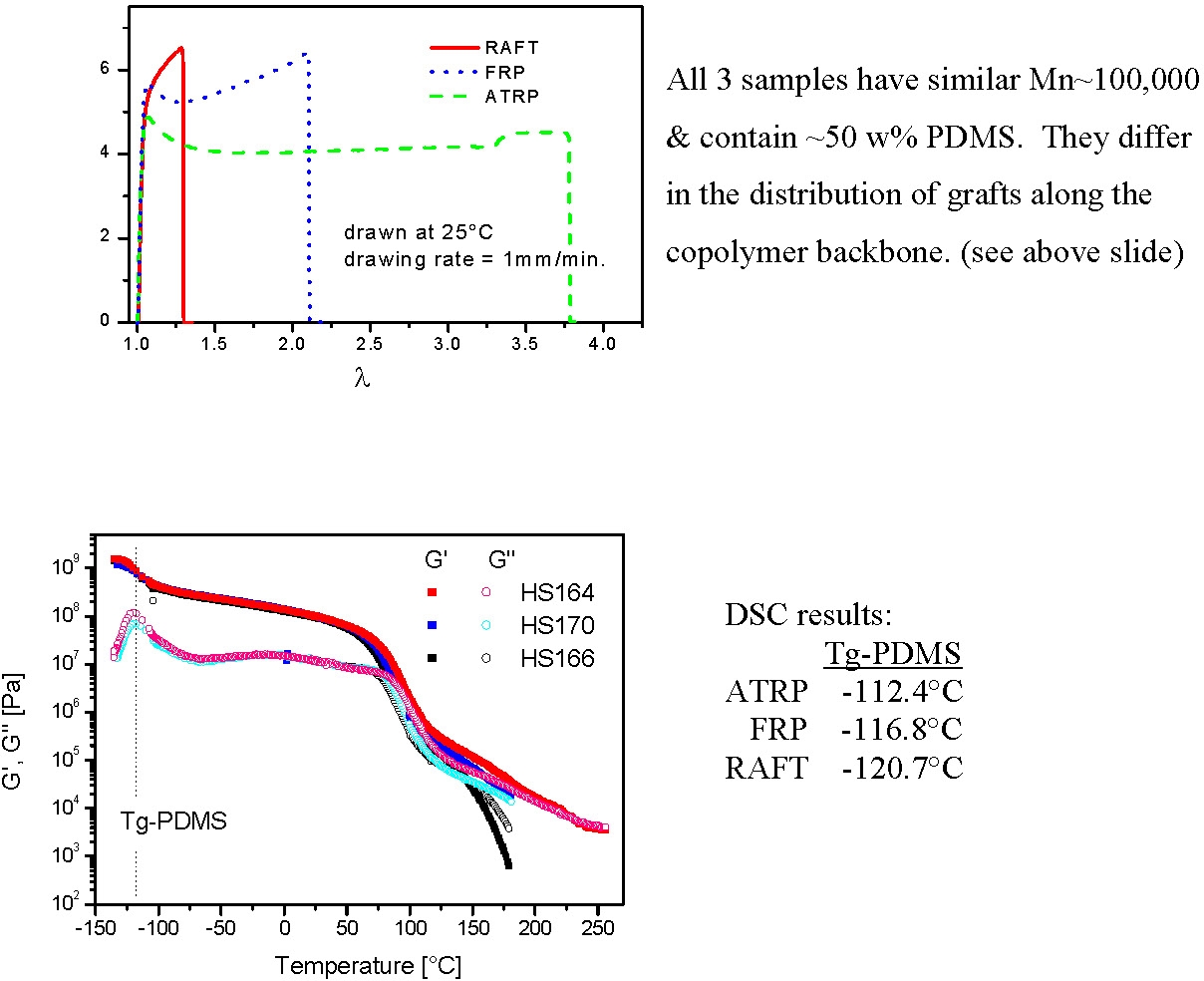
As shown in the following cartoons, copolymerization by conventional FRP led to the preparation of copolymers that displayed a broad distribution of chain length and composition due to the continuous change in the instantaneous feed ratios of the comonomers remaining in the reaction medium and the effect of continuous initiation/growth/termination of the copolymerization throughout the reaction. At the other extreme ATRP performed in 30% xylene solution at 90 0C utilizing a compatible macroinitiator resulted in preparation of a uniform comb-like graft copolymer due to similar reactivity of both comonomers, (r ~1) in the diluted reaction solution. However, conducting ATRP or RAFT in only 3% xylene generated gradient copolymers, due to monomer feed variation throughout the reaction because of the impact of viscosity on macromonomer reactivity resulting in a higher reactivity ratio for MMA, (r~2), compared to the macromonomer.
The following schematics illustrate the differences in the microstructure and also the dramatic affect the differences in microstructure had on tensile properties.
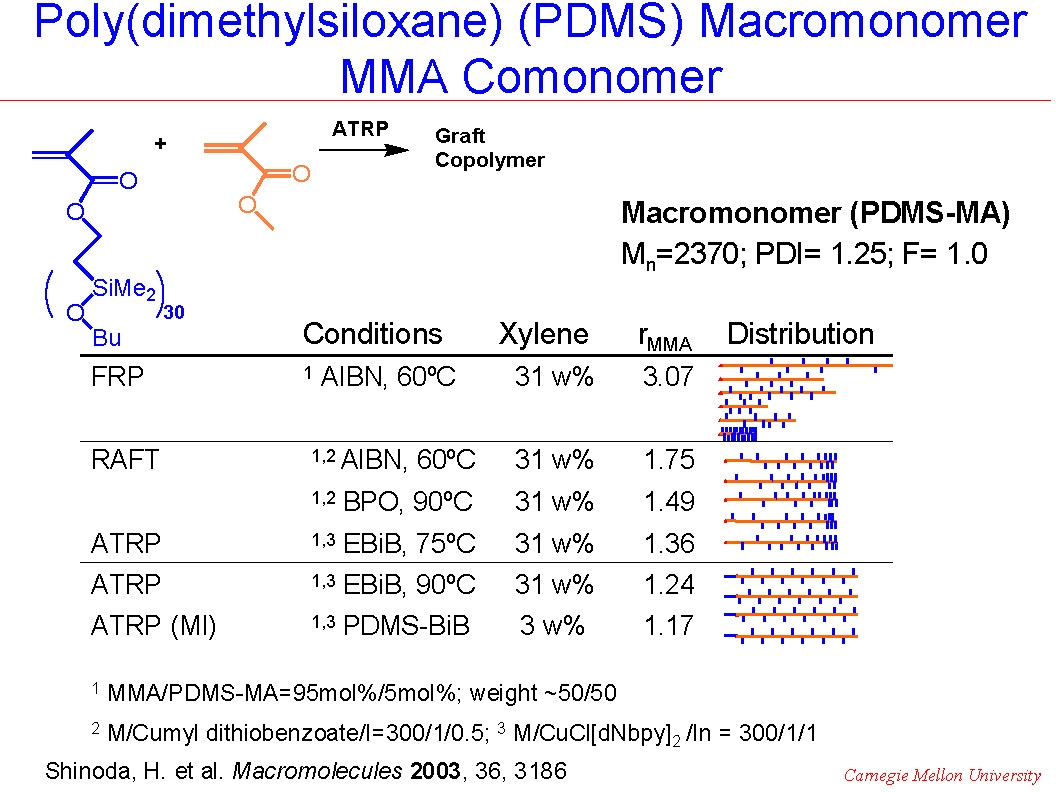
The tapered, or gradient graft copolymer has a tensile elongation of only ~30%, compared to ~120% for the irregular non-homogeneous product formed by FRP, or the~280% for a regular comb copolymer.(4) These dramatic differences in physical properties occur even though all three copolymers have approximately the same overall composition (~50/50) and the same overall molecular weight Mw~100,000.
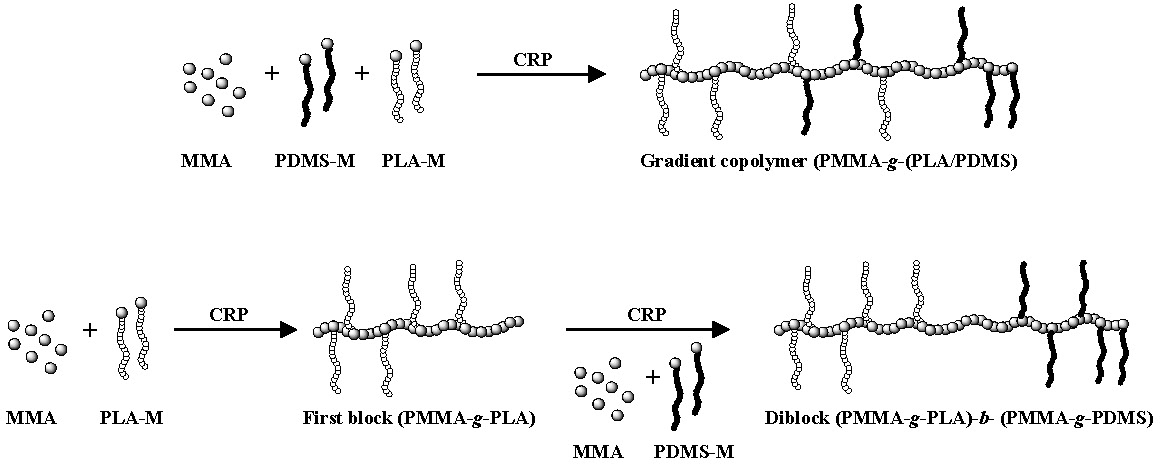
Topological control can also be extended to heterograft copolymers(5) where graft copolymers with grafts of different composition can be prepared in a one step or two step CRP grafting through procedure with two different macromonomers forming copolymers with different gradient distributions of grafts along the backbone of the copolymer. In the following set of schematics the different reactivity of a PLA macromonomer and a PDMS macromonomer are shown to result in the preparation of a gradient copolymer with a gradient of both macromonomers along the polymer backbone. The PLA-M is preferentially incorporated while the PDMS-M is incorporated later in the copolymerization process forming a copolymer with a higher concentration of PLA grafts at one chain end and a higher concentration of PDMS grafts at the other. This situation is amplified when a two step copolymerization is conducted.
ATRP of two poly(ethylene oxide) (PEO) macromonomers, with different degrees of polymerization (DPn) and different end groups, was conducted in solution via the grafting through method.(6) Selection of a PEO methacrylate with a methyl end-group (PEOMeMA, DPPEO = 23) and a PEO acrylate end-capped by a Phenyl ring (PEOPhA, DPPEO = 4) for the copolymerization led to a spontaneous gradient of PEO grafts along the copolymer backbone. A spontaneous gradient copolymer was formed because of significantly different reactivity of the two PEO macromonomers. The resulting copolymer has PEOMeMA at one end of the polymer chain, gradually changing through hetero-sequences to predominately PEOPhA grafts at the other chain end. An increase in the initial feed ratio of PEO acrylate reduced the rate of change in the shape of the gradient. Amorphous-crystalline structure in the copolymers was demonstrated by DSC and WAXS. The mechanical measurements of copolymers consisting of an amorphous PEOPhA and crystallizable PEOMeMA segments indicated elastomeric properties in the range of a soft rubber (G' approximatly 104 Pa, G' >> G'').
Grafting from:
Graft copolymer with a poly(vinyl chloride) backbone were prepared by two approaches, one was preparation of a macroinitiator by copolymerization of vinyl chloride with a monomer containing an ATRP initiating group, vinyl chloroacetate.(7) The other was to graft from inherent structural defects in poly(vinyl chloride.(8) Either approach can be considered to be a viable approach to self plasticized PVC.(9)
Another example of a graft copolymer prepared by ATRP using the "grafting from" approach is the formation of a polyethylene-g-poly(n-butyl acrylate) copolymer which used a polyethylene macroinitiator with distributed α-bromoisobutyrate functionality. The copolymers were prepared using a less active BA6TREN based ligand that was soluble in the reaction medium to controllably polymerize butyl acrylate in a solution of the polyethylene macroinitiator.(10,11) Depending on the mole ratio of the segments such a material could find application as an impact modifier for polyethylene, a compatibilizer in polyethylene blends, as a surfactant or as a surface modifier to provide better adhesive properties. The macroinitiator was prepared by copolymerization of ethylene with undecenol using a zirconocene based catalyst. The pendant hydroxyl groups were esterified ml 2-bromopropionyl bromide for a butyl acrylate grafting from reaction.
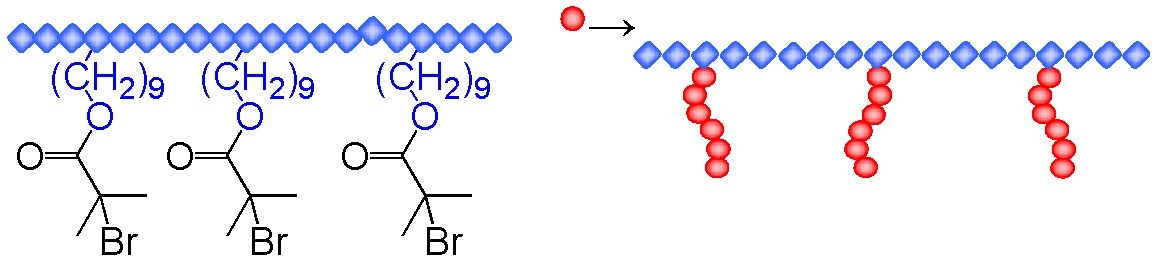
There is strong incompatibility between the components and the final copolymer, containing 32-67% butyl acrylate units, showed both clear melting and crystallization peaks attributable to polyethylene and a glass transition from the poly(butyl acrylate). At lower mole fractions of poly(butyl acrylate) the matrix was based on polyethylene and displayed good extensibility as well as molecular and structural orientability while the material with a poly(butyl acrylate) matrix displayed a loss of resistance to tensile deformation. Below the glass transition the of the PBA phase the PBA seems to play a role of reinforcement in the PE matrix, so that the modulus of the material can even exceed the values determined for the pure polyethylene. Above the glass transition of the PBA, the soft phase of grafts influences the modulus of the polymers to a degree dependent on composition.
A strong effect of the presence of the grafted chains was also seen in the molten state in which the grafted polymers behave as elastomers with remarkably higher modulus than that determined for the entangled polyethylene. The observed behavior should be related to the two phase structure of the system with PBA micro-domains dispersed in the PE matrix and networked by the PE backbone chains.
As shown below the PE-g-MMA demonstrate good performance as a compatibilizer between the two homopolymers when the blends were studied by TEM.(12)

Grafting to:
As discussed on page 7 graft copolymers can also be prepared by "grafting to" reactions.(13-15) While grafting to is perhaps the oldest procedure for preparation of graft copolymers in free radical chemistry, e.g., impact resistant polystyrene, properties of materials prepared by grafting to in a controlled polymerization process has not yet gained a significant amount of attention.
REFERENCES
(1) Boerner, H. G.; Matyjaszewski, K. Macromol. Symp. 2002, 177, 1.
(2) Inceoglu, S.; Olugebefola, S. C.; Acar, M. H.; Mayes, A. M. Designed Monomers and Polymers 2004, 7, 181-189.
(3) Shinoda, H.; Miller, P. J.; Matyjaszewski, K. Macromolecules 2001, 34, 3186-3194.
(4) Shinoda, H.; Matyjaszewski, K.; Okrasa, L.; Mierzwa, M.; Pakula, T. Macromolecules 2003, 36, 4772-4778.
(5) Lutz, J.-F.; Jahed, N.; Matyjaszewski, K. Journal of Polymer Science, Part A: Polymer Chemistry 2004, 42, 1939-1952.
(6) Neugebauer, D.; Zhang, Y.; Pakula, T. Journal of Polymer Science, Part A: Polymer Chemistry 2006, 44, 1347-1356.
(7) Paik, H. J.; Gaynor, S. G.; Matyjaszewski, K. Macromol. Rapid Commun. 1998, 19, 47-52.
(8) Percec, V.; Asgarzadeh, F. J. Polym. Sci., Part A: Polym. Chem. 2001, 39, 1120-1135.
(9) Spanswick, J.; Branstetter, E. A.; Huber, W. F., Jr. ACS Symp. Ser. 2000, 768, 427-440.
(10) Okrasa, L.; Pakula, T.; Inoue, Y.; Matyjaszewski, K. Colloid and Polymer Science 2004, 282, 844-853.
(11) Inoue, Y.; Matyjaszewski, K. Journal of Polymer Science, Part A: Polymer Chemistry 2004, 42, 496-504.
(12) Inoue, Y.; Matsugi, T.; Kashiwa, N.; Matyjaszewski, K. Macromolecules 2004, 37, 3651-3658.
(13) Gao, H.; Matyjaszewski, K. J. Am. Chem. Soc. 2007, 129, 6633-6639.
(14) Van Camp, W.; Germonpre, V.; Mespouille, L.; Dubois, P.; Goethals, E. J.; Du Prez, F. E. Reactive & Functional Polymers 2007, 67, 1168-1180.
(15) Tsarevsky, N. V.; Bencherif, S. A.; Matyjaszewski, K. Macromolecules 2007, 40, 4439-4445.