
Dr. Andreas Pfenning
Associate Professor, Computational Biology, Neuroscience Institute and Biomedical Engineering
- Gates Hillman Center Room 7711
Gates Hillman Center Room 7711
Carnegie Mellon University
5000 Forbes Avenue
Pittsburgh, PA 15213
Education
- B.S., Computer Science, Carnegie Mellon University, 2006
- Ph.D., Computational Biology, Duke University, 2012
- Post-Doctoral, Computations Biology, MIT, 2015
Bio
Andreas Pfenning is a tenured Associate Professor in the Computational Biology Department in the School of Computer Science and a member of the Neuroscience Institute. Previously, he completed a joint postdoctoral position in Computer Science at MIT (CSAIL) and the Genetics Department at Harvard Medical School. He received his Ph.D. in Computational Biology from Duke University and his bachelor’s degree in computer science from Carnegie Mellon University.
Andreas has won several additional accolades including a Sloan Foundation research fellowship, an NSF Career Award, and an American Federation for Aging Research grant. He also received an Avenir award, a grant given by the National Institute of Drug Abuse to fund forward-thinking research by new investigators in substance use disorders. His research on the evolution of speech received an HPCWire award for the Best Use of AI Methods for Augmenting High Performance Computing Applications. He currently the chair of comparative genomics working group of the vertebrate genome project, leveraging hundreds of high-quality genomes to better understand the principles of genome evolution and human health.
Research
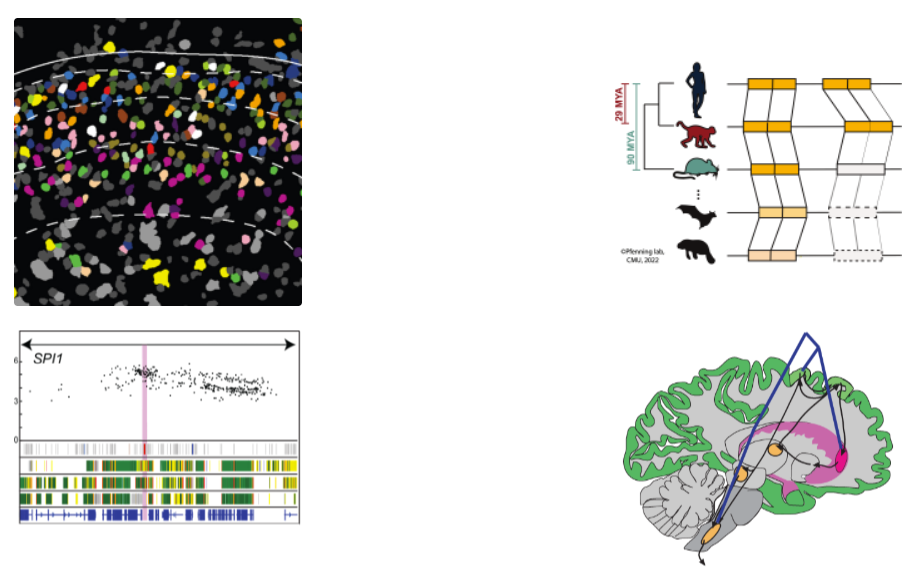
The Pfenning laboratory studies how genetic and epigenetic variation influences human brain disorders and evolution of complex behaviors. One of the biggest challenges in understanding the brain is cell type heterogeneity: even a small piece of brain tissue can contain dozens of different cell types that interact to play a variety of roles across different behaviors. The Pfenning laboratory disentangles that heterogeneity by combining AI approaches with genomic technology development. Convolutional neural networks and other machine learning models provide the means to predict how genetic differences with the human population and across species will influence gene regulation across neural cell types. Those computational models are informed by in vivo rodent experiments conducted by the Pfenning laboratory, including epigenomics, in vivo massively parallel reporter assays, and spatial transcriptomics. Their research has been published in top venues, including Cell, Nature, and a series of four Science articles applying comparative genomics to study genetic variation across species and within the human population.
The Pfenning laboratory has integrated machine learning and experimental genomic approaches to study a variety of brain disorders. New virus-based experimental genomic technologies developed by the Pfenning laboratory, which have now been adopted in over 80 laboratories around the world, was used collect genomic data on different neural cell types. That dataset, along with publicly available data, has been used by the Pfenning laboratory to train AI models that predict the roles that different cell types play in Alzheimer’s disease, addiction, and Parkinson’s disease, and aging. Those predictions have been validated and refined using in vivo massively parallel reporter assays and spatial transcriptomic approaches that can measure how synthetic fragments of DNA function across different brain regions and cell types. Moving forward, the Pfenning laboratory is using AI and spatial transcriptomics to develop targeted therapeutics for chronic pain, Parkinson’s disease, and other brain disorders.
In parallel to the research centered on variation within the human population, the Pfenning laboratory is combining machine learning and comparative genomics to study how complex traits and behaviors have evolved. Methods to measure conservation or convergent evolution at regulatory elements have overwhelmingly relied on models that examine whether individual nucleotides are conserved. This assumption doesn’t match recent observations that regulatory function can be maintained in specific cell types across species despite extremely low conservation at the individual nucleotide level. To better study regulatory element evolution, we developed TACIT, in which we examine whether the cell type-specific function of region of the genome is predicted to be conserved by convolutional neural network models. Using our approach, we have been able to identify dozens of regions of the genome that are associated with the evolution of brain size and vocal learning behavior, the basis of human speech.
Research Interests: Machine learning, artificial intelligence, genetics, genomics, neurological disorders, Alzheimer’s disease
Awards and Recognition
-
NSF Career Award, 2021