Computer Simulation Breaks Virus Apart To Learn How it Comes Together
By Jocelyn Duffy / 412-268-9982 / jhduffy@andrew.cmu.eduResearchers led by Carnegie Mellon University physicist Markus Deserno and University of Konstanz (Germany) chemist Christine Peter have developed a computer simulation that crushes viral capsids. By allowing researchers to see how the tough shells break apart, the simulation provides a computational window for looking at how viruses and proteins assemble.
The study is published in the October issue of The European Physical Journal Special Topics.
“The concept of breaking something to see how it’s made isn’t new. It’s what’s being done at particle accelerators and in materials science labs worldwide — not to mention by toddlers who break their toys to see what’s inside,” said Deserno, a professor in the Department of Physics and member of the department’s Biological Physics Initiative. “With a simulation we can build the virus, crush it and see what happens at a very high level of resolution.”
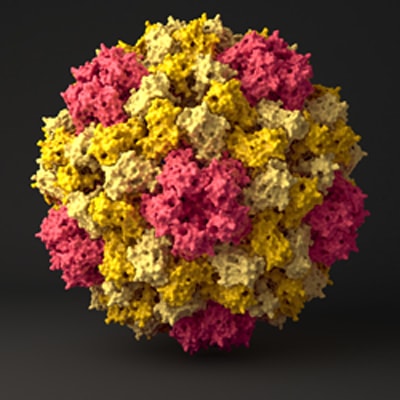
Computer simulation of Cowpea Chlorotic Mottle Virus capsid.
Credit: Venkatramanan Krishnamani
Viral capsids, the protein shells that encapsulate and transport the viral genome, are one of nature’s strongest nanocontainers. The shells are made when copies of capsid proteins spontaneously come together and assemble into a round, geometric shell. Understanding how these proteins come together to form capsids may help researchers to make similar nanocontainers for a variety of uses, including targeted drug delivery. Additionally, the simulation could fill a void for virologists, allowing them to study the stages of viral assembly that they aren’t able to see experimentally.
Studying the self-assembly of viral capsids is difficult. Most viruses are too small — about 30 to 50 nanometers — and the capsid proteins come together too rapidly for their assembly to be seen using traditional microscopy. As an alternative, Deserno and colleagues thought a better way to learn about capsid assembly might be to see what happens when an already formed capsid breaks apart.
To do this, Deserno and colleagues created a coarse-grained model of the Cowpea Chlorotic Mottle Virus (CCMV) capsid. In the simulation, they applied forces to the capsid and viewed how it responded to those forces. Their model is based on the MARTINI force field, a commonly used coarse-grained model, with an added stabilizing network within the individual proteins that compensated for the model’s shortcomings in stabilizing a protein’s folding geometry.
The CCMV capsid is made up of 180 identical proteins. In assembly, the proteins first form pairs, called dimers, and those dimers then join together at interfaces. While the proteins are the same, the interfaces can be different. At some locations on the capsid, five proteins meet; at others, six meet. In the simulation, the researchers found that when force was applied to the capsid, the capsid would start to fracture at the hexametric interfaces first, indicating that those protein-protein contacts were weaker than those at the pentametric interfaces. In contrast, the pentametric contacts never broke. Since stronger connections assemble first and weaker ones assemble later, the researchers can use this information to begin to recreate how the capsid formed.
In the simulation, the researchers also found a likely explanation for a strange structural feature found in the CCMV capsid. At the center of the hexametric association site, the tail ends of the six proteins come together and form a beta barrel. Beta barrels are coiled secondary protein structures. The researchers believe they act to provide further late-stage stabilization to the weaker hexametric interfaces.
The research was funded by the Volkswagen Foundation. Additional authors include: Venkatramanan Krishnamani, former CMU postdoc now at the University of Iowa; and Christoph Globisch and Christine Peter, University of Konstanz, Germany, and Max-Planck Institute for Polymer Research, Mainz, Germany.