| |
|
|
Electrostatic Interaction Controls Binding Orientation of the HIV-1 Matrix Domain to Acidic Membranes
Hirsh Nanda,† Siddhartha A. K. Datta,* Frank Heinrich,§,† Mathias Lösche,§,† Joseph E. Curtis,† Alan Rein,* and Susan Krueger†
†National Institute of Standards and Technology, Gaithersburg, MD; *HIV Drug Resistance Program, National Cancer Institute, Frederick, MD; §Carnegie Mellon University, Pittsburgh, PA
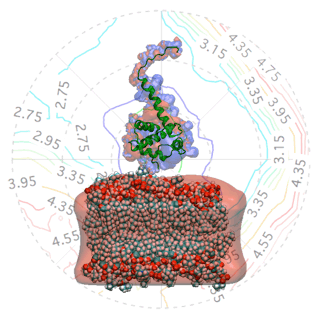
The Gag polyprotein of human immunodeficiency virus type-1 (HIV-1) plays a central role in mediating viral assembly in an infected host cell [1]. Gag is composed of several functional domains, the matrix (MA), capsid (CA), nucleocapsid (NC) and p6. To initiate budding of a daughter viral particle, the N-terminal MA domain targets and binds the plasma membrane where subsequently viral assembly occurs [2]. The atomic structure of the isolated MA domain has been solved using NMR spectroscopy [3].
Several molecular mechanisms are implicated in the membrane association of the MA domain and its selectivity for the plasma membrane.
-
A myristate anchor is co-translationally attached to the N-terminus of MA and is believed to insert into the membrane [4,5].
-
MA engages in specific binding to the phosphoinositoldiphosphate PI(4,5)P 2 which is enriched in the interior layer of the plasma membrane [3,6]. MA binding to PIP 2 is thought to trigger a conformational change of the protein by which the myristate, which is sequestered inside MA in buffer, is exposed thereby making it available for membrane binding [3].
-
A patch of basic amino acid residues on the protein surface may interact electrostatically with anionic membrane lipids [7,8].
 To study the contributions of these interactions to the recruiting of MA to the plasma membrane, we initiated a study of unmyristoylated –myrMA to artificial membranes (tethered bilayer lipid membranes, tBLMs) that bear similar charge density as plasma membranes but lacks their full complexity. In a first key experiment, reported here, we were particularly interested to determine to what extent electrostatic interactions alone can drive the association of MA with such tBLMs. To study the contributions of these interactions to the recruiting of MA to the plasma membrane, we initiated a study of unmyristoylated –myrMA to artificial membranes (tethered bilayer lipid membranes, tBLMs) that bear similar charge density as plasma membranes but lacks their full complexity. In a first key experiment, reported here, we were particularly interested to determine to what extent electrostatic interactions alone can drive the association of MA with such tBLMs.
|
Tethered Bilayer Membranes (tBLMs)
We devised synthetic thiolipids in which a thiol is separated from a hydrophobic lipid anchor by a hydrophilic oligo(ethyleneoxide) spacer [9,10]. Self-assembled monolayers (SAMs) of these compounds are formed on gold-coated substrates and completed with phospholipids. In this work, we used a mixture of DMPC and DMPG (7:3). In the completed surface-supported membrane, the lipids are decoupled from the gold surface by the spacer, as a nanometer-thin, stable aqueous layer is formed between the membrane and the substrate. Neutron reflectometry (NR) and electrical impedance spectroscopy (EIS) show complete, electrically-sealing tBLMs, that can cover large areas (cm2) and are stable for days [11].
To ensure hydration of the submembrane space requires "backfilling", i.e., spacing out the grafted thiolipids in-plane with a diluent, such as 2-mercaptoethanol (βME). tBLMs can be completed with a range of lipids, saturated or unsaturated, charged or zwitterionic. |
Electrostatic Membrane Binding of –myrMA
The unmyristoylated MA domain binds to anionic lipid bilayers with high affinity. We studied the structure of the resulting surface architecture with neutron reflectometry (NR).
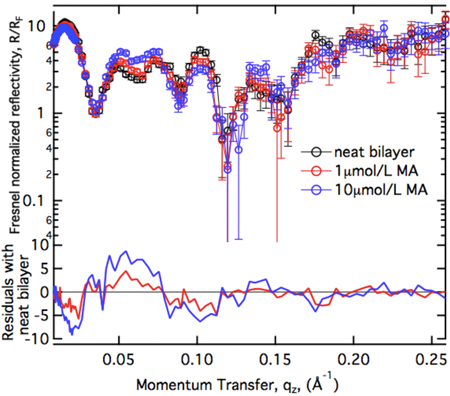
The uniform increase of the residuals as a function of protein concentration suggests that MA binds in a uniform monolayer at the membrane which increases in packing density as more protein becomes available for binding. Models with progressively higher complexity have been applied to interprete the NR spectra quantitatively.
|
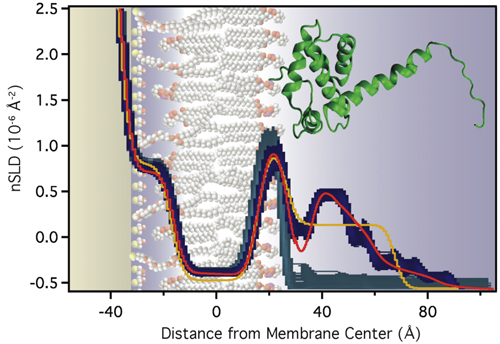
Molecular-Scale Interpretation of the NR Results
The modeling results suggest that electrostatic interactions alone are sufficient to target the protein to the membrane in the correct orientation for PIP2 binding and myristoyl anchoring. If we assume that the NMR structure of MA is a valid description of the protein structure at the membrane, the protein's penetration depth into the bilayer derived from the model fit to the data lets us explore which amino acids penetrate into the lipid headgroups and lead to the stable association of the protein with the membrane, even in absence of the myristoyl anchor or membrane-bound PIP2. We thus group the surface exposed basic AA residues into three groups: (1) AAs who interact intimately with the lipid headgroups (with the distribution of their Cα atoms shown in yellow below). (2) AAs that interact marginally in the assumed orientation of the protein (shown in green). These residues might interact more strongly if the protein orientation was somewhat tilted from the assumed one. (3) AAs that do not interact with the membrane (shown in red).
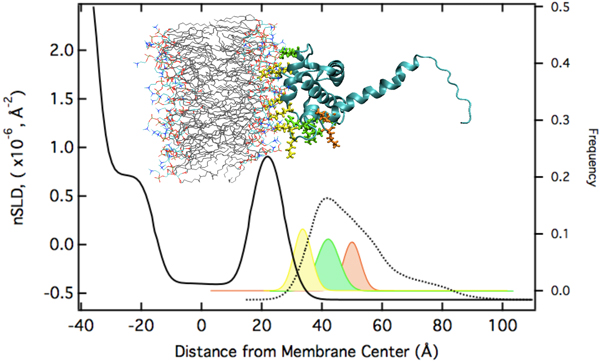
|
MA Orientation on the Membrane
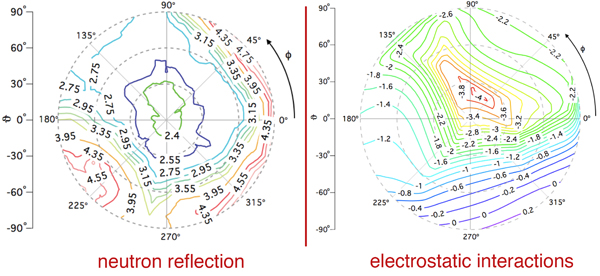
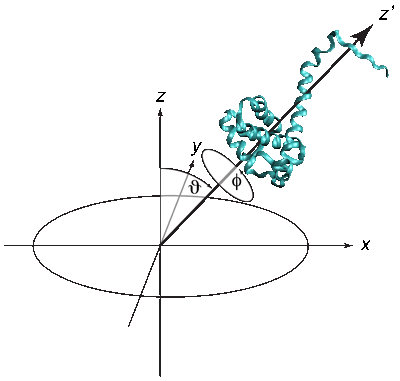 The quality of the NR data allowed us to refine the orientation dependence of the model. We parameterized orientation in terms of Euler angles (only two angles, ϑ and φ need to be considered, as the third Euler angle is irrelevant due to the cylindrical symmetry of the problem). Both the best-fit to the NR data (left) and a simplified electrostatic energy calculation (right) suggest that the protein may be tilted by ≈ 20° from the canonical orientation inferred from the trimeric X-ray structure. The quality of the NR data allowed us to refine the orientation dependence of the model. We parameterized orientation in terms of Euler angles (only two angles, ϑ and φ need to be considered, as the third Euler angle is irrelevant due to the cylindrical symmetry of the problem). Both the best-fit to the NR data (left) and a simplified electrostatic energy calculation (right) suggest that the protein may be tilted by ≈ 20° from the canonical orientation inferred from the trimeric X-ray structure.
|
Hierarchical Levels of Protein-Membrane Interactions may Determine Binding of MA to Plasma Membrane
The results obtained in this work and prior knowledge of the protein structure and of functional aspects of MA binding to bilayers suggest the following hierarchical model:
- Electrostatic interactions funnel MA to the membrane in the correct orientation for tighter binding in levels 2 and 3. These interactions are relatively long-range but weak, however, they are apparently sufficient to predispose the protein for subsequent binding interactions by imposing the correct orientation of the protein at the interface for the next steps.
- Specific binding of the MA domain to PI(4,5)P2 targets Gag to the correct membrane, i.e., the plasma membrane, and releases the myristate from its sequestered state, presumably by activating an allosteric switch. The exposed myristate is then available for
- insertion due to hydrophobic interactions into the lipid membrane thus finalizing the stable anchoring of the protein at the target membrane.
|
The research described here benefited greatly from neutron research facilities provided by the National Institute of Standards and Technology at the NIST Center for Neutron Research and the usage of the Beowulf Linux cluster located at the NIH. It is supported by the U.S. Department of Commerce (MSE 70NANB8H009) and the Intramural Research rogram of the NIH. |
References
[1] Ganser-Pornillos, B. K.; Yeager, M.; Sundquist, W. I. Curr. Opin. Struct. Biol. 2008, 18, 203.
[2] Bukrinskaya, A. Virus Res. 2007, 124, 1.
[3] Saad, J. S.; Miller, J.; Tai, J.; Kim, A.; Ghanam, R. H.; Summers, M. F. Proc. Natl. Acad. Sci. USA 2006, 103, 11364.
[4] Göttlinger, H. G.; Sodroski, J. G.; Haseltine, W. A. Proc. Natl. Acad. Sci. USA 1989, 86, 5781.
[5] Spearman, P.; Horton, R.; Ratner, L.; Kuli-Zade, I. J. Virol. 1997, 71, 6582.
[6] Ono, A.; Ablan, S. D.; Lockett, S. J.; Nagashima, K.; Freed, E. O. Proc. Natl. Acad. Sci. USA 2004, 101, 14889.
[7] Zhou, W. J.; Parent, L. J.; Wills, J. W.; Resh, M. D. J. Virol. 1994, 68, 2556.
[8] Ono, A.; Orenstein, J. M.; Freed, E. O. J. Virol. 2000, 74, 2855.
[9] McGillivray, D. J.; Valincius, G.; Vanderah D. J.; Febo-Ayala, W.; Woodward, J. T.; Heinrich, F.; Kasianowicz, J. J.; Lösche, M. Biointerphases 2007, 2, 21.
[10] Heinrich F.; Ng, T.; Vanderah, D. J.; Shekhar, P.; Mihailescu, M.; Nanda, H.; Lösche, M. Langmuir 2009 25, 4219.
[11] Valincius, G.; McGillivray, D. J.; Febo-Ayala, W.; Vanderah, D. J.; Kasianowicz, J. J.; Lösche, M. J. Phys. Chem. B 2006, 110, 10213.
[12] Hill, C. P.; Worthylake, D.; Bancroft, D. P.; Christensen, A. M.; Sundquist, W. I. Proc. Natl. Acad. Sci. USA 1996, 93, 3099.
[13] McGillivray, D. J.; Valincius, G.; Heinrich, F.; Robertson, J. W. F.; Vanderah. D. J.; Febo-Ayala, W. Ignatjev, I.; Lösche, M.; Kasianowicz, J. J. Biophys. J. 2009, 96, 1547. |
|
|
|

