| |
|
|
A New Spin on Alzheimer's Disease:
A Membrane-Mediated Mechanism for Amyloid-β Toxicity?
Frank Heinrich,§,† Charles G. Glabe,* Yuri Sokolov,* James E. Hall,* Gintaras Valincius,** and Mathias Lösche§,†
§Carnegie Mellon University, Pittsburgh, PA; †NIST Center f. Neutron Research, Gaithersburg, MD; *University of California at Irvine, CA; **Institute of Biochemistry, Vilnius, Lithuania
Alzheimer’s Disease (AD) is the most common cause of dementia among people over 60. An irreversible, progressive brain disease that slowly destroys memory and thinking skills, Alzheimer’s affects as many as 5 million Americans and 27 million people worldwide. In the brain of a person with Alzheimer’s, changes may begin as early as 10 to 20 years before obvious symptoms, such as memory loss, become apparent.
While the basic pathology of Alzheimer’s was discovered a century ago, and substantial research efforts have since been devoted, it remains unclear where or how the disease begins. At late stages of the disease, insoluble plaques composed of the misfolded chains of a protein fragment called amyloid β (Aβ) accumulate in the patient’s brain. However, recent research has indicated that smaller Aβ aggregates, which are soluble in the intercellular fluid and may form at an earlier stage, are the most toxic form of the peptide. These toxic Aβ oligomers may infest the brain long before large-scale brain damage becomes apparent. It has been established that the particles interact strongly with cell membranes, possibly causing nerve cells to leak. The leaky membranes are unable to maintain the difference of ion concentration between the inside and outside of cells that is essential to information processing, and consequently to central nervous system function. |
Soluble Amyloid-β Oligomers Interact Strongly with Membranes
Because cleavage of the Amyloid Precursor Protein (APP) may occur at different positions, different variants of the Aβ peptide are released in the brain. While the major component of plaques is the Aβ(1-40), the slightly more hydrophobic Aβ(1-42) appears to be the more toxic variant. These peptide are widely accepted to play a key role in the etiology of Alzheimer’s disease. However, the actual mechanism of cell toxicity of Aβ remains unclear and is the object of intensive research.
While amyloid fibrils, mainly composed of Aβ(1-40) and Aβ(1-42), are the most conspicuous morphological structures observed in the brains of deceased patients, there is more and more experimental evidence that the Aβ oligomers is the aggregation form that inflicts the most damage [1,2]. In fact, small soluble amyloidic oligomers deriving from a variety of proteins in very different tissues have recently been suspected to lead to pathologies in many amyloid diseases, such as AD (Aβ), Parkinson's (α-synuclein), Huntington's (huntingtin), type-2 diabetes (islet amyloid polypeptide, IAPP), as well as prion diseases. Because the affected tissues show this broad distribution, it has been speculated that all these misfolded peptide oligomers target a common morphological structure: the cell membrane. One of the most important questions with respect to the molecular-scale details that give rise to AD is then, How do misfolded Aβ peptides inflict damage on neuronal membranes? |
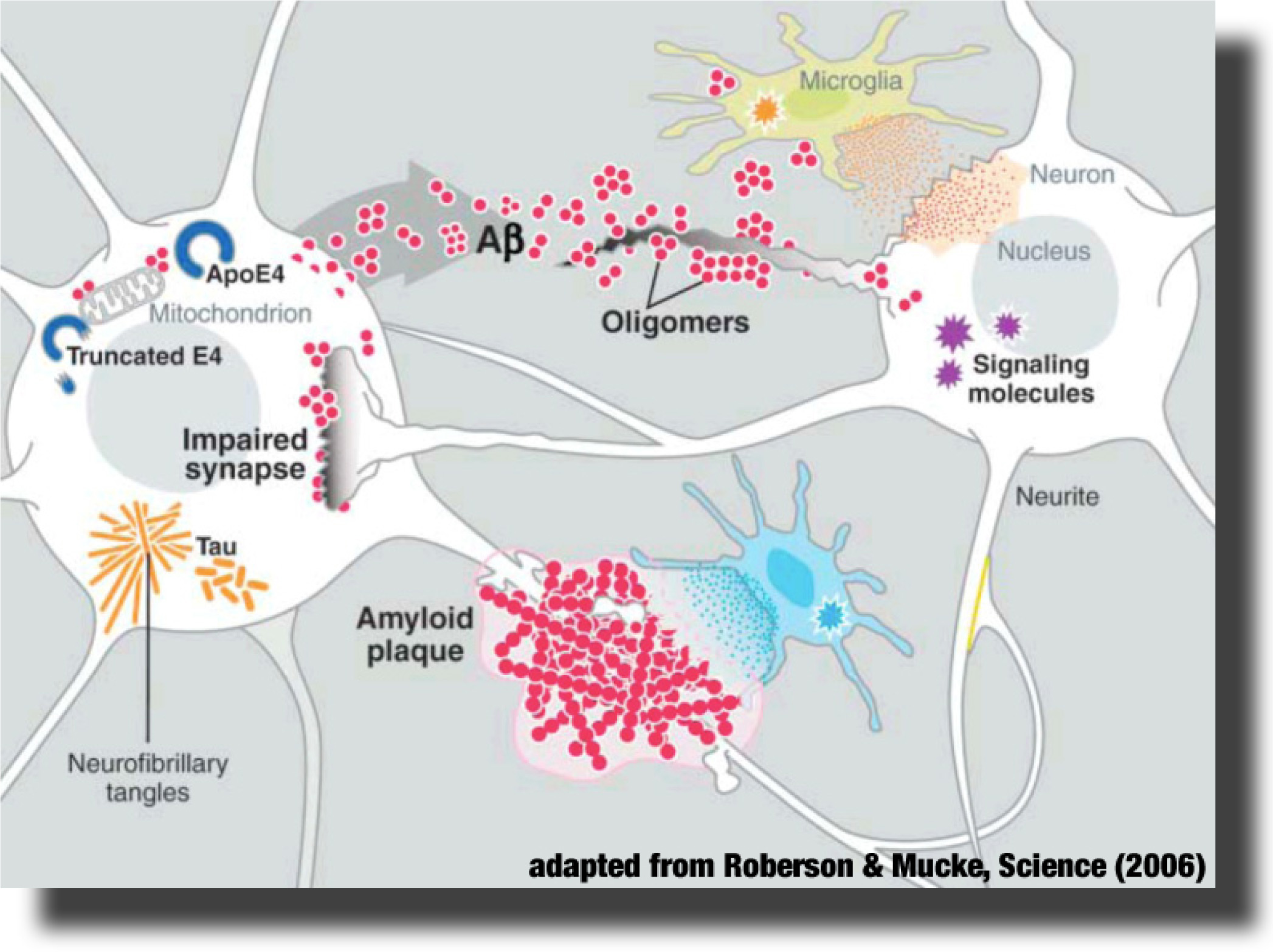 The Alzheimer’s peptide Aβ is a natural cleavage product released within neuronal cells. If misfolded, Aβ forms distinct structures that are prone to aggregation, first into oligomers, then into progressively larger structures called amyloid plaques. It is now believed that Aβ oligomers are the toxic form of the peptide that may impair basic neuronal processes, such as the communication between the nerve cells, long before clinical symptoms become obvious. However, molecular details of these early processes remain largely unknown. The Alzheimer’s peptide Aβ is a natural cleavage product released within neuronal cells. If misfolded, Aβ forms distinct structures that are prone to aggregation, first into oligomers, then into progressively larger structures called amyloid plaques. It is now believed that Aβ oligomers are the toxic form of the peptide that may impair basic neuronal processes, such as the communication between the nerve cells, long before clinical symptoms become obvious. However, molecular details of these early processes remain largely unknown.
|
We have recently initiated a research program, supported by the National Institute of Aging and the American Health Assistance Foundation, aimed at investigation of the molecular-scale etiology of Alzheimer's, as far as it relates to Aβ impact on membranes. This research program brings together a wide range of expertise from studies in silico (peptide and membrane simulations by the Deserno group) via experimental studies of tethered bilayer lipid membranes (tBLMs) as artificial membrane systems (Lösche and Valincius groups) to biophysical studies of free-standing membranes and electrophysiological measurements in cell cultures (Hall group). The Glabe group supports the project with peptide chemistry and immunological agents [1].
|
Aβ Oligomers do not Form Pores but Reorganize the Lipid Matrix at Low Peptide Concentration
In an initial study, we investigated the hypothesis that Aβ forms protein channels in membranes [3,4], similar to those formed by bacterial toxins. Indeed, Aβ oligomers increase the conductivity of artificial membranes and cell membranes in cultured cells dramatically at micromolar peptide concentrations [5–7]. However, when we examine microscopic details of the changes in membrane properties that lead to this increase, we find strong evidence that protein pore formation is not responsible for the breakdown of the membrane barrier to ion transfer [7].
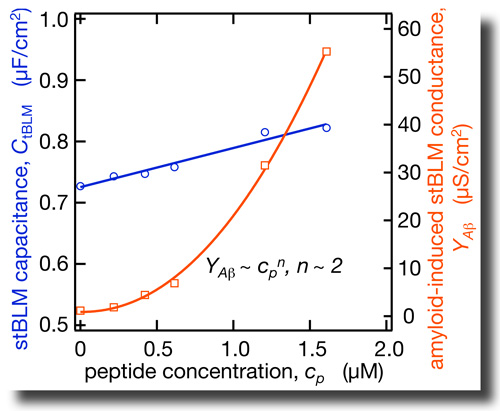 By combining EIS with neutron reflection measurements we determined the dielectric properties of the hydrophobic core in tBLMs on an absolute scale. In as-prepared tBLMs, the dielectric constant is ε = 2.2 and 2.8 for bilayers completed with DPhyPC and DOPC, respectively. NR shows that Aβ(1–42) oligomers insert deeply into the membrane. This membrane association of the peptide is accompanied with a strong increase in the dielectric constant in the bilayer. The dependence of these effects on peptide concentration is nonlinear. By combining EIS with neutron reflection measurements we determined the dielectric properties of the hydrophobic core in tBLMs on an absolute scale. In as-prepared tBLMs, the dielectric constant is ε = 2.2 and 2.8 for bilayers completed with DPhyPC and DOPC, respectively. NR shows that Aβ(1–42) oligomers insert deeply into the membrane. This membrane association of the peptide is accompanied with a strong increase in the dielectric constant in the bilayer. The dependence of these effects on peptide concentration is nonlinear. |
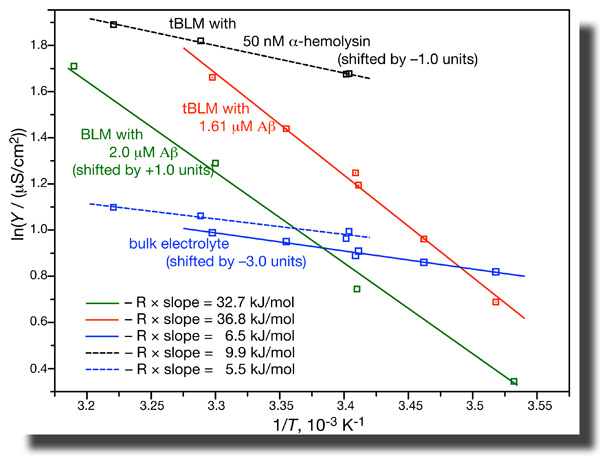 While all these results are consistent with pore formation by the peptide, further evidence is not. EIS of tBLMs determines the conductivity in the bulk buffer phase and across the membrane independently in one measurement. If we do such measurements as a function of temperature, we can determine the activation energies of ion transport from the Arrhenius plots. It turns out that these activation energies are grossly different for ion transport across the membrane as induced by Aβ(1–42) oligomers and by α-hemolysin. Whereas ion transport through the αHL channel has an activation energy of ~ 10 kJ/mol, the value we measure for transport through Aβ-affected membranes are closer to 35 kJ/mol [7]. This is a heck of a difference! While all these results are consistent with pore formation by the peptide, further evidence is not. EIS of tBLMs determines the conductivity in the bulk buffer phase and across the membrane independently in one measurement. If we do such measurements as a function of temperature, we can determine the activation energies of ion transport from the Arrhenius plots. It turns out that these activation energies are grossly different for ion transport across the membrane as induced by Aβ(1–42) oligomers and by α-hemolysin. Whereas ion transport through the αHL channel has an activation energy of ~ 10 kJ/mol, the value we measure for transport through Aβ-affected membranes are closer to 35 kJ/mol [7]. This is a heck of a difference!
Furthermore, if one evaluates the average increase of the dielectric constant upon peptide incorporation into the bilayer and tries to match this increase with the observed increase in bilayer conductivity for ions, it is realized that these quantities do not match – the conductivity increase is much larger than expected from the change in ε. Only if we assume that the peptide changes the local fabric of the membrane, thus increasing ε locally by a great amount while intervening membrane patches remain unaffected can we quantitatively match conductivity and dielectric properties of the membrane affected by Aβ [7].
The conclusion from this work is that Aβ(1–42) oligomers are very unlikely to perforate neuronal membranes, particularly at peptide concentrations that are in the physiological range, i.e., orders of magnitude lower than the micromolar concentrations used in our study.
|
The research in this project has been published in G. Valincius et al., Biophys. J. (2008), 4845, ref. [7] below. It is supported by the AHAF (A2008-307) and the NIH (1 P01 AG032131). This project benefits greatly from neutron neutron research facilities provided by the National Institute of Standards and Technology at the NIST Center for Neutron Research.
References
[1] Kayed R.; Head E.; Thompson J. L.; McIntire T. M.; Milton S. C.; Cotman C. W.; Glabe C. G. (2003) Science 300, 486.
[2] Lesné S.; Koh M. T.; Kotilinek L.; Kayed R.; Glabe C. G.; Yang A.; Gallagher M.; Ashe K. H. (2006), Nature 440, 352.
[3] Arispe N.; Rojas E.; Pollard H. B. (1993), Proc. Natl. Acad. Sci. USA 90, 567.
[4] Lin H.; Bhatia R.; Lal R. (2001), FASEB J. 15, 2433.
[5] Kayed R.; Sokolov Y.; Edmonds B.; McIntire T. M.; Milton S. C.; Hall J. E.; Glabe C. G. (2004), J. Biol. Chem. 279, 46363.
[6] Sokolov Y.; Kozak J. A.; Kayed R.; Chanturiya A.; Glabe C. G.; Hall J. E. (2006), J. Gen. Physiol. 128, 637.
[7] Valincius G.; Heinrich F.; Budvytyte R.; Vanderah D. J.; McGillivray D. J.; Sokolov Y.; Hall J. E.; Lösche M. (2008), Biophys. J. 95, 4845. |
|
|
|


