| |
 |
|
Publications
Regular Articles |
| |
2020 |
| |
The Dynamics of Active Nematic Defects on the Surface of a Sphere
Yi-Heng Zhang, Markus Deserno, and Zhan-Chun Tu
arXiv.org > cond-mat > arXiv:2006.02947.
 A nematic liquid crystal confined to the surface of a sphere exhibits topological defects of total charge +2 due to the topological constraint. In equilibrium, the nematic field forms four +1/2 defects, located at the corners of a regular tetrahedron inscribed within the sphere, since this minimizes the Frank elastic energy. If additionally the individual nematogens exhibit self-driven directional motion, the resulting active system creates large-scale flow that drives it out of equilibrium. In particular, the defects now follow complex dynamic trajectories which, depending on the strength of the active forcing, can be periodic (for weak forcing) or chaotic (for strong forcing). In this paper we derive an effective particle theory for this system, in which the topological defects are the degrees of freedom, whose exact equations of motion we subsequently determine. Numerical solutions of these equations confirm previously observed characteristics of their dynamics and clarify the role played by the time dependence of their global rotation. We also show that Onsager's variational principle offers an exceptionally transparent way to derive these dynamical equations, and we explain the defect mobility at the hydrodynamics level. A nematic liquid crystal confined to the surface of a sphere exhibits topological defects of total charge +2 due to the topological constraint. In equilibrium, the nematic field forms four +1/2 defects, located at the corners of a regular tetrahedron inscribed within the sphere, since this minimizes the Frank elastic energy. If additionally the individual nematogens exhibit self-driven directional motion, the resulting active system creates large-scale flow that drives it out of equilibrium. In particular, the defects now follow complex dynamic trajectories which, depending on the strength of the active forcing, can be periodic (for weak forcing) or chaotic (for strong forcing). In this paper we derive an effective particle theory for this system, in which the topological defects are the degrees of freedom, whose exact equations of motion we subsequently determine. Numerical solutions of these equations confirm previously observed characteristics of their dynamics and clarify the role played by the time dependence of their global rotation. We also show that Onsager's variational principle offers an exceptionally transparent way to derive these dynamical equations, and we explain the defect mobility at the hydrodynamics level.
|
|
Probing Nanoparticle/Membrane Interactions by Combining Amphiphilic Diblock Copolymer Assembly and Plasmonics
Amelie H.R.Koch, Svenja Morsbach, Tristan Bereau,Gaëtan Lévêque, Hans-Jürgen Butt, Markus Deserno, Katharina Landfester, and George Fytas
J. Phys. Chem. B 124, 742–750 (2020).
 Understanding the interactions between nanoparticles (NPs) and boundaries of cells is crucial both for their toxicity and therapeutic applications. Besides specific receptor-mediated endocytosis of surface-functionalized NPs,
passive internalization is prompted by relatively unspecific parameters, such as particle size and charge. Based on theoretical treatments, adhesion to and bending of the cell membrane can induce NP wrapping. Experimentally, powerful tools are needed to selectively probe possible membrane-NP motifs at very dilute conditions and avoid dye labeling. In this work, we employ surface resonance-enhanced dynamic light scattering, surface plasmon resonance, electron microscopy, and plasmonic AuNPs and polymersomes. We distinguish three different interaction scenarios at nanomolar concentrations by tuning the surface charge of AuNPs and rationalize these events by balancing vesicle bending and electrostatic/van der Waals AuNP and vesicle adhesion. The clarification of the physical conditions under which nanoparticles passively translocate across membranes can aid in the rational design of drugs that cannot exploit specific modes of cellular uptake and also elucidates physical properties that render nanoparticles in the environment particularly toxic. Understanding the interactions between nanoparticles (NPs) and boundaries of cells is crucial both for their toxicity and therapeutic applications. Besides specific receptor-mediated endocytosis of surface-functionalized NPs,
passive internalization is prompted by relatively unspecific parameters, such as particle size and charge. Based on theoretical treatments, adhesion to and bending of the cell membrane can induce NP wrapping. Experimentally, powerful tools are needed to selectively probe possible membrane-NP motifs at very dilute conditions and avoid dye labeling. In this work, we employ surface resonance-enhanced dynamic light scattering, surface plasmon resonance, electron microscopy, and plasmonic AuNPs and polymersomes. We distinguish three different interaction scenarios at nanomolar concentrations by tuning the surface charge of AuNPs and rationalize these events by balancing vesicle bending and electrostatic/van der Waals AuNP and vesicle adhesion. The clarification of the physical conditions under which nanoparticles passively translocate across membranes can aid in the rational design of drugs that cannot exploit specific modes of cellular uptake and also elucidates physical properties that render nanoparticles in the environment particularly toxic.
|
|
Spontaneous curvature, differential stress, and bending modulus of asymmetric lipid membranes
Amirali Hossein and Markus Deserno
Biophys. J. 118, 624–642 (2020).
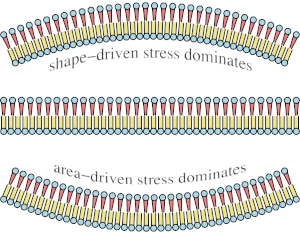 Lipid bilayers can exhibit asymmetric states in which the physical characteristics of one leaflet differ from those of the other. This most visibly manifests in a different lipid composition, but it can also involve opposing lateral stresses in each leaflet that combine to an overall vanishing membrane tension. Here we use theoretical modeling and coarse-grained simulation to explore the interplay between a compositional asymmetry and a nonvanishing differential stress. Minimizing the total elastic energy leads to a preferred spontaneous curvature that balances torques due to both bending moments and differential stress, with sometimes unexpected consequences. For instance, asymmetric flat bilayers, whose specific areas in each leaflet are matched to those of corresponding tensionless symmetric flat membranes, still exhibit a residual differential stress, because the conditions of vanishing area strain and vanishing bending moment differ. We also measure the curvature rigidity of asymmetric bilayers and find that a sufficiently strong differential stress, but not compositional asymmetry alone, can increase the bending modulus. The likely cause is a stiffening of the compressed leaflet, which appears to be related to its gel transition, but not identical with it. We finally show that the impact of cholesterol on differential stress depends on the relative strength of elastic and thermodynamic driving forces: if cholesterol solvates equally well in both leaflets, it will redistribute to cancel both leaflet tensions almost completely; but if its partitioning free energy prefers one leaflet over the other, the resulting distribution bias may even create differential stress. Since cells keep most of their lipid bilayers in an asymmetric nonequilibrium steady state, our findings suggests that biomembranes are elastically more complex than previously thought: besides a spontaneous curvature they might also exhibit significant differential stress, which could strongly affect their curvature energetics. Lipid bilayers can exhibit asymmetric states in which the physical characteristics of one leaflet differ from those of the other. This most visibly manifests in a different lipid composition, but it can also involve opposing lateral stresses in each leaflet that combine to an overall vanishing membrane tension. Here we use theoretical modeling and coarse-grained simulation to explore the interplay between a compositional asymmetry and a nonvanishing differential stress. Minimizing the total elastic energy leads to a preferred spontaneous curvature that balances torques due to both bending moments and differential stress, with sometimes unexpected consequences. For instance, asymmetric flat bilayers, whose specific areas in each leaflet are matched to those of corresponding tensionless symmetric flat membranes, still exhibit a residual differential stress, because the conditions of vanishing area strain and vanishing bending moment differ. We also measure the curvature rigidity of asymmetric bilayers and find that a sufficiently strong differential stress, but not compositional asymmetry alone, can increase the bending modulus. The likely cause is a stiffening of the compressed leaflet, which appears to be related to its gel transition, but not identical with it. We finally show that the impact of cholesterol on differential stress depends on the relative strength of elastic and thermodynamic driving forces: if cholesterol solvates equally well in both leaflets, it will redistribute to cancel both leaflet tensions almost completely; but if its partitioning free energy prefers one leaflet over the other, the resulting distribution bias may even create differential stress. Since cells keep most of their lipid bilayers in an asymmetric nonequilibrium steady state, our findings suggests that biomembranes are elastically more complex than previously thought: besides a spontaneous curvature they might also exhibit significant differential stress, which could strongly affect their curvature energetics.
See also the New and Notabe "Differential or Curvature Stress? Modus Vivendi" by Edward Lyman and Alexander Sodt.: Biophys. J. 118, 535–537 (2020). You can also watch a talk on Youtube about this paper.
|
|
2019 |
| |
Mechanical Properties of Lipid Bilayers: A Note on the Poisson Ratio
M. Mert Terzi, Markus Deserno, and John F. Nagle
Soft Matter 15, 9085–9092 (2019).
 We investigate the Poisson ratio ν of fluid lipid bilayers, i.e., the question how area strains compare to the changes in membrane thickness (or, equivalently, volume) that accompany them. We first examine existing experimental results on the area- and volume compressibility of lipid membranes. Analyzing them within the framework of linear elasticity theory for homogeneous thin fluid sheets leads us to conclude that lipid membrane deformations are to a very good approximation volume-preserving, with a Poisson ratio that is likely about 3% smaller than the common soft matter limit ν=1/2. These results are fully consistent with atomistic simulations of a DOPC membrane at varying amount of applied lateral stress, for which we instead deduce ν by directly comparing area- and volume strains. To assess the problematic assumption of transverse homogeneity, we also define a depth-resolved Poisson ratio ν(z) and determine it through a refined analysis of the same set of simulations. We find that throughout the membrane's thickness, ν(z) is close to the value derived assuming homogeneity, with only minor variations of borderline statistical significance.
We investigate the Poisson ratio ν of fluid lipid bilayers, i.e., the question how area strains compare to the changes in membrane thickness (or, equivalently, volume) that accompany them. We first examine existing experimental results on the area- and volume compressibility of lipid membranes. Analyzing them within the framework of linear elasticity theory for homogeneous thin fluid sheets leads us to conclude that lipid membrane deformations are to a very good approximation volume-preserving, with a Poisson ratio that is likely about 3% smaller than the common soft matter limit ν=1/2. These results are fully consistent with atomistic simulations of a DOPC membrane at varying amount of applied lateral stress, for which we instead deduce ν by directly comparing area- and volume strains. To assess the problematic assumption of transverse homogeneity, we also define a depth-resolved Poisson ratio ν(z) and determine it through a refined analysis of the same set of simulations. We find that throughout the membrane's thickness, ν(z) is close to the value derived assuming homogeneity, with only minor variations of borderline statistical significance.
|
|
A Consistent Quadratic Curvature-Tilt Theory For Fluid Lipid Membranes
M. Mert Terzi, Muhammed F. Ergüder, and Markus Deserno
J. Chem. Phys. 151, 164108 (2019).
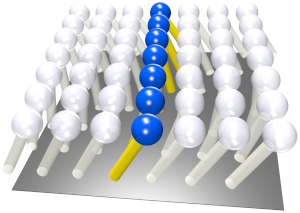 The tilt of a lipid molecule describes the deviation of its orientation away from the local normal of its embedding membrane. Tilt is the subleading degree of freedom after a membrane's geometry, and it becomes relevant at scales comparable to lipid bilayer thickness. Building on earlier work by Hamm and Kozlov (Eur. Phys. J. E 3, 323 (2000)), who envisioned lipid membranes as thin pre-stressed fluid elastic films, and Terzi and Deserno (J. Chem. Phys. 147, 084702 (2017)), who discovered a new coupling term between splay and tilt divergence, we construct a theory of membrane elasticity that is quadratic in geometry and tilt and complete at order 1/length2. We show that a general and consistent treatment of both lateral and transverse depth-dependent shear stresses creates several contributions to the elastic energy density, of which only a subset had previously been identified. Apart from the well-known penalty of lipid twist (the curl of tilt), these terms generate no qualitatively new phenomenology, but they quantitatively revise the connections between the moduli of a tilt-curvature theory and its underlying microscopic foundation. In particular, we argue that the monolayer Gaussian curvature modulus κm', widely believed to be equal to the second moment of the transmonolayer stress profile, acquires a second contribution from lipid twist, which is always negative. This could resolve the long-standing conundrum that many measured values of κm' appeared to have a sign that violates basic stability considerations. We also show that the previously discovered novel coupling between splay and tilt divergence is not simply proportional to κm' but acquires its own splay-tilt coupling modulus, κst. We explore the predictions of our theory for various elastic moduli and their mutual interrelations, and use an extensive set of existing atomistic molecular dynamics simulations for twelve different lipid types to collectively reason about such predictions. We find that bending rigidities are captured fairly well by existing theories, while reliable predictions for local moduli, especially the splay-tilt coupling modulus, remain challenging.
The tilt of a lipid molecule describes the deviation of its orientation away from the local normal of its embedding membrane. Tilt is the subleading degree of freedom after a membrane's geometry, and it becomes relevant at scales comparable to lipid bilayer thickness. Building on earlier work by Hamm and Kozlov (Eur. Phys. J. E 3, 323 (2000)), who envisioned lipid membranes as thin pre-stressed fluid elastic films, and Terzi and Deserno (J. Chem. Phys. 147, 084702 (2017)), who discovered a new coupling term between splay and tilt divergence, we construct a theory of membrane elasticity that is quadratic in geometry and tilt and complete at order 1/length2. We show that a general and consistent treatment of both lateral and transverse depth-dependent shear stresses creates several contributions to the elastic energy density, of which only a subset had previously been identified. Apart from the well-known penalty of lipid twist (the curl of tilt), these terms generate no qualitatively new phenomenology, but they quantitatively revise the connections between the moduli of a tilt-curvature theory and its underlying microscopic foundation. In particular, we argue that the monolayer Gaussian curvature modulus κm', widely believed to be equal to the second moment of the transmonolayer stress profile, acquires a second contribution from lipid twist, which is always negative. This could resolve the long-standing conundrum that many measured values of κm' appeared to have a sign that violates basic stability considerations. We also show that the previously discovered novel coupling between splay and tilt divergence is not simply proportional to κm' but acquires its own splay-tilt coupling modulus, κst. We explore the predictions of our theory for various elastic moduli and their mutual interrelations, and use an extensive set of existing atomistic molecular dynamics simulations for twelve different lipid types to collectively reason about such predictions. We find that bending rigidities are captured fairly well by existing theories, while reliable predictions for local moduli, especially the splay-tilt coupling modulus, remain challenging.
|
|
2018 |
| |
The role of scaffold reshaping and disassembly in dynamin driven membrane fission
Martina Pannuzzo, Zachary A. McDargh, and Markus Deserno
eLife 2018;7:e39441.
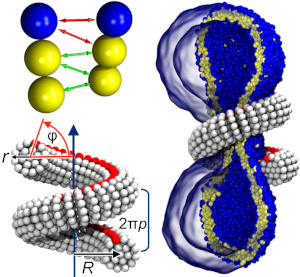 The large GTPase dynamin catalyzes membrane fission in eukaryotic cells, but despite three decades of experimental work, competing and partially conflicting models persist regarding some of its most basic actions. Here we investigate the mechanical and functional consequences of dynamin scaffold shape changes and disassembly with the help of a geometrically and elastically realistic simulation model of helical dynamin-membrane complexes. Beyond changes of radius and pitch, we emphasize the crucial role of a third functional motion: an effective rotation of the filament around its longitudinal axis, which reflects alternate tilting of dynamin's PH binding domains and creates a membrane torque. We also show that helix elongation impedes fission, hemifission is reached via a small transient pore, and coat disassembly assists fission. Our results have several testable structural consequences and help to reconcile mutual conflicting aspects between the two main present models of dynamin fission-the two-stage and the constrictase model.
The large GTPase dynamin catalyzes membrane fission in eukaryotic cells, but despite three decades of experimental work, competing and partially conflicting models persist regarding some of its most basic actions. Here we investigate the mechanical and functional consequences of dynamin scaffold shape changes and disassembly with the help of a geometrically and elastically realistic simulation model of helical dynamin-membrane complexes. Beyond changes of radius and pitch, we emphasize the crucial role of a third functional motion: an effective rotation of the filament around its longitudinal axis, which reflects alternate tilting of dynamin's PH binding domains and creates a membrane torque. We also show that helix elongation impedes fission, hemifission is reached via a small transient pore, and coat disassembly assists fission. Our results have several testable structural consequences and help to reconcile mutual conflicting aspects between the two main present models of dynamin fission-the two-stage and the constrictase model.
|
|
Optimal coarse-grained site selection in elastic network models of biomolecules
Patrick Diggins IV, Changjiang Liu, Markus Deserno, and Raffaello Potestio
J. Chem. Theory Comput., in press (2018).
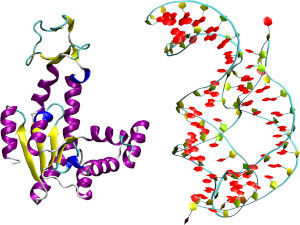 Elastic network models, simple structure-based representations of biomolecules where atoms interact via short-range harmonic potentials, provide great insight into a molecule's internal dynamics and mechanical properties at extremely low computational cost. Their efficiency and effectiveness have made them a pivotal instrument in the computer-aided study of proteins and, since a few years, also of nucleic acids. In general, the coarse-grained sites, i.e.i> those effective force centres onto which the all-atom structure is mapped, are constructed based on intuitive rules: a typical choice for proteins is to retain only the Cα atoms of each amino acid. However, a mapping strategy relying only on the atom type and not the local properties of its embedding can be suboptimal compared to a more careful selection. Here we present a strategy in which the subset of atoms, each of which is mapped onto a unique coarse-grained site of the model, is selected in a stochastic search aimed at optimising a cost function. The latter is taken to be a simple measure of the consistency between the harmonic approximation of an elastic network model and the harmonic model obtained through exact integration of the discarded degrees of freedom. The method is applied to two representatives of structurally very different types of biomolecules: the protein Adenylate kinase and the RNA molecule adenine riboswitch. Our analysis quantifies the substantial impact that an algorithm-driven selection of coarse-grained sites can have on a model's properties.
Elastic network models, simple structure-based representations of biomolecules where atoms interact via short-range harmonic potentials, provide great insight into a molecule's internal dynamics and mechanical properties at extremely low computational cost. Their efficiency and effectiveness have made them a pivotal instrument in the computer-aided study of proteins and, since a few years, also of nucleic acids. In general, the coarse-grained sites, i.e.i> those effective force centres onto which the all-atom structure is mapped, are constructed based on intuitive rules: a typical choice for proteins is to retain only the Cα atoms of each amino acid. However, a mapping strategy relying only on the atom type and not the local properties of its embedding can be suboptimal compared to a more careful selection. Here we present a strategy in which the subset of atoms, each of which is mapped onto a unique coarse-grained site of the model, is selected in a stochastic search aimed at optimising a cost function. The latter is taken to be a simple measure of the consistency between the harmonic approximation of an elastic network model and the harmonic model obtained through exact integration of the discarded degrees of freedom. The method is applied to two representatives of structurally very different types of biomolecules: the protein Adenylate kinase and the RNA molecule adenine riboswitch. Our analysis quantifies the substantial impact that an algorithm-driven selection of coarse-grained sites can have on a model's properties.
|
|
The 2018 Biomembrane Curvature and Remodeling Roadmap
Patricia Bassereau, Rui Jin, Tobias Baumgart, Markus Deserno, Rumiana Dimova, Vadim A. Frolov, Pavel V. Bashkirov, Helmut Grubmüller, Reinhard Jahn, H. Jelger Risselada, Ludger Johannes, Michael M. Kozlov, Reinhard Lipowsky, Thomas J. Pucadyil, Wade F. Zeno, Jeanne C. Stachowiak, Dimitrios Stamou, Artu Breuer, Line Lauritsen, Camille Simon, Cécile Sykes, Gregory A. Voth, and Thomas R. Weikl
J. Phys. D: Appl. Phys. 51, 343001 (2018).
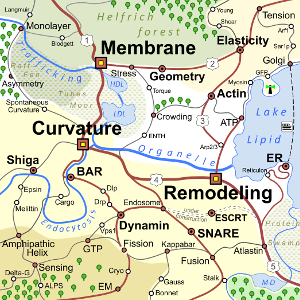 The importance of curvature as a structural feature of biological membranes has been recognized for many years and has fascinated scientists from a wide range of different backgrounds. On the one hand, changes in membrane morphology are involved in a plethora of phenomena involving the plasma membrane of eukaryotic cells, including endo- and exocytosis, phagocytosis and filopodia formation. On the other hand, a multitude of intracellular processes at the level of organelles rely on generation, modulation, and maintenance of membrane curvature to maintain the organelle shape and functionality. The contribution of biophysicists and biologists is essential for shedding light on the mechanistic understanding and quantification of these processes.
The importance of curvature as a structural feature of biological membranes has been recognized for many years and has fascinated scientists from a wide range of different backgrounds. On the one hand, changes in membrane morphology are involved in a plethora of phenomena involving the plasma membrane of eukaryotic cells, including endo- and exocytosis, phagocytosis and filopodia formation. On the other hand, a multitude of intracellular processes at the level of organelles rely on generation, modulation, and maintenance of membrane curvature to maintain the organelle shape and functionality. The contribution of biophysicists and biologists is essential for shedding light on the mechanistic understanding and quantification of these processes.
Given the vast complexity of phenomena and mechanisms involved in the coupling between membrane shape and function, the road towards the next major steps in accomplishing an exhaustive understanding in this research area can be unclear. The 2018 Biomembrane curvature and remodeling roadmap of the Journal of Physics D: Applied Physics addresses this need for clarity and is intended to provide guidance both for students who just enter the field as well as established scientists who would like to improve their orientation within this fascinating area.
|
|
Responsive Behavior of a Branched-Chain Polymer Network: a Molecular Dynamics Study
Martina Pannuzzo, Robert D. Tilton, and Markus Deserno
Soft Matter 14, 6485-6495 (2018).
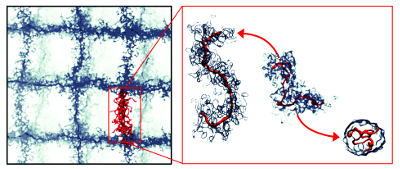 Smart polymer hydrogels, which can undergo structural and volume phase transitions in response to external stimuli, have gained much attention for their widespread technological applications. Compared to linear polymers, branched chains offer more extensive opportunities to rationally design functional materials, since they permit more extensive structural tunability—for instance by adjusting the balance between hydrophobic and hydrophilic units, the grafting fraction of backbone monomers, or the side chain length, topology, and solubility. Here we conduct coarse-grained molecular dynamics simulations to assess how well generic physical principles capture this complex interplay of tuning parameters, specifically when building networks from complex branched chains with a hydrophobic backbone. Swollen chains collapse upon reducing side chain solubility, length, and grafting density, but neither the sharpness of this transition nor its dynamic range, if measured via chain extension, depends monotonically on these parameters. Networks comprising such chains are more swollen and exhibit even sharper transitions, but their higher responsiveness goes along with a swelling ratio that falls behind that of single chains. Smart polymer hydrogels, which can undergo structural and volume phase transitions in response to external stimuli, have gained much attention for their widespread technological applications. Compared to linear polymers, branched chains offer more extensive opportunities to rationally design functional materials, since they permit more extensive structural tunability—for instance by adjusting the balance between hydrophobic and hydrophilic units, the grafting fraction of backbone monomers, or the side chain length, topology, and solubility. Here we conduct coarse-grained molecular dynamics simulations to assess how well generic physical principles capture this complex interplay of tuning parameters, specifically when building networks from complex branched chains with a hydrophobic backbone. Swollen chains collapse upon reducing side chain solubility, length, and grafting density, but neither the sharpness of this transition nor its dynamic range, if measured via chain extension, depends monotonically on these parameters. Networks comprising such chains are more swollen and exhibit even sharper transitions, but their higher responsiveness goes along with a swelling ratio that falls behind that of single chains.
|
|
Dynamin's helical geometry does not destabilize membranes during fission
Zachary A. McDargh and Markus Deserno
Traffic 19, 328–335 (2018).
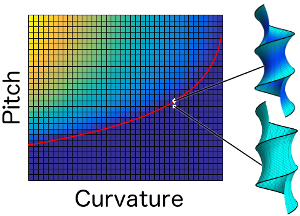 It is now widely accepted that dynamin mediated fission is a fundamentally mechanical process: dynamin undergoes a GTP-dependent conformational change, constricting the neck between two compartments, somehow inducing their fission. However, the exact connection between dynamin’s conformational change and the scission of the neck is still unclear. In this paper, we re-evaluate the suggestion that a change in the pitch or radius of dynamin’s helical geometry drives the lipid bilayer through a mechanical instability, similar to a well-known phenomenon occurring in soap films. We find that, contrary to previous claims, there is no such instability. This lends credence to an alternative model, in which dynamin drives the membrane up an energy barrier, allowing thermal fluctuations to take it into the hemifission state.
It is now widely accepted that dynamin mediated fission is a fundamentally mechanical process: dynamin undergoes a GTP-dependent conformational change, constricting the neck between two compartments, somehow inducing their fission. However, the exact connection between dynamin’s conformational change and the scission of the neck is still unclear. In this paper, we re-evaluate the suggestion that a change in the pitch or radius of dynamin’s helical geometry drives the lipid bilayer through a mechanical instability, similar to a well-known phenomenon occurring in soap films. We find that, contrary to previous claims, there is no such instability. This lends credence to an alternative model, in which dynamin drives the membrane up an energy barrier, allowing thermal fluctuations to take it into the hemifission state.
|
|
2017 |
| |
Novel Tilt–Curvature Coupling in Lipid Membranes
M. Mert Terzi and Markus Deserno
J. Chem. Phys. 147, 084702 (2017).
 On mesoscopic scales lipid membranes are well described by continuum theories whose main ingredients are the curvature of a membrane's reference surface and the tilt of its lipid constituents. In particular, Hamm and Kozlov [Eur. Phys. J. E 3, 323 (2000)] have shown how to systematically derive such a tilt-curvature Hamiltonian, based on the elementary assumption of a thin fluid elastic sheet experiencing internal lateral pre-stress. Performing a dimensional reduction, they not only derive the basic form of the effective surface Hamiltonian, but also express its emergent elastic couplings as trans-membrane moments of lower-level material parameters. In the present paper we argue, though, that their derivation unfortunately missed a coupling term between curvature and tilt. This term arises because, as one moves along the membrane, the curvature-induced change of transverse distances contributes to the area strain—an effect that was believed to be small, but nevertheless ends up contributing at the same (quadratic) order as all other terms in their Hamiltonian. We illustrate the consequences of this amendment by deriving the monolayer and bilayer Euler-Lagrange equations for the tilt, as well as the power spectra of shape, tilt, and director fluctuations. A particularly curious aspect of our new term is that its associated coupling constant is the second moment of the lipid monolayer's lateral stress profile—which within this framework is equal to the monolayer Gaussian curvature modulus, κm. On the one hand, this implies that many theoretical predictions now contain a parameter that is poorly known (because the Gauss-Bonnet theorem limits access to the integrated Gaussian curvature); on the other hand, the appearance of κm outside of its Gaussian curvature provenance opens opportunities for measuring it by more conventional means, for instance by monitoring a membrane's undulation spectrum at short scales.
On mesoscopic scales lipid membranes are well described by continuum theories whose main ingredients are the curvature of a membrane's reference surface and the tilt of its lipid constituents. In particular, Hamm and Kozlov [Eur. Phys. J. E 3, 323 (2000)] have shown how to systematically derive such a tilt-curvature Hamiltonian, based on the elementary assumption of a thin fluid elastic sheet experiencing internal lateral pre-stress. Performing a dimensional reduction, they not only derive the basic form of the effective surface Hamiltonian, but also express its emergent elastic couplings as trans-membrane moments of lower-level material parameters. In the present paper we argue, though, that their derivation unfortunately missed a coupling term between curvature and tilt. This term arises because, as one moves along the membrane, the curvature-induced change of transverse distances contributes to the area strain—an effect that was believed to be small, but nevertheless ends up contributing at the same (quadratic) order as all other terms in their Hamiltonian. We illustrate the consequences of this amendment by deriving the monolayer and bilayer Euler-Lagrange equations for the tilt, as well as the power spectra of shape, tilt, and director fluctuations. A particularly curious aspect of our new term is that its associated coupling constant is the second moment of the lipid monolayer's lateral stress profile—which within this framework is equal to the monolayer Gaussian curvature modulus, κm. On the one hand, this implies that many theoretical predictions now contain a parameter that is poorly known (because the Gauss-Bonnet theorem limits access to the integrated Gaussian curvature); on the other hand, the appearance of κm outside of its Gaussian curvature provenance opens opportunities for measuring it by more conventional means, for instance by monitoring a membrane's undulation spectrum at short scales.
|
|
2016 |
| |
Constriction by Dynamin: elasticity vs. adhesion
Zachary A. McDargh, Pablo Vázquez-Montejo, Jemal Guven, and Markus Deserno
Biophys. J. 111, 2470–2480 (2016).
 Any cellular fission process is completed when the neck connecting almost-separate membrane compartments is severed. This crucial step is somehow accomplished by proteins from the dynamin family, which polymerize into helical spirals around such necks. Much research has been devoted to elucidating the specifics of that “somehow”, but despite no shortage of ideas, the question is not settled. Pictorially obvious notions of “strangling” or “pushing” are difficult to render in mechanically precise terms. Moreover, since dynamin is a GTPase, it is tempting to speculate that it has a motor activity that assists the necessary severing “action”, but again the underlying mechanics is not obvious. We believe the difficulty to be the mechanically nontrivial nature of confining elastic filaments onto curved surfaces, for which efficient methods to conceptualize the associated forces and torques have only recently appeared. Here we investigate the implications of a conceptually simple yet mechanically challenging model: consider an elastic helical filament confined to a surface mimicking the neck between two membrane compartments, which we assume to take the shape of a catenoid. What can we say about the expected length of such adsorbed filaments, their shapes, and the forces they exert, as a function of the key parameters in the model? While real dynamin is surely more complex, we consider such a minimal model to be the indispensable baseline. Without knowing what such a model can and cannot explain, it is difficult to justify more complex mechanisms, or understand the constraints under which this machinery evolved in the first place.
Any cellular fission process is completed when the neck connecting almost-separate membrane compartments is severed. This crucial step is somehow accomplished by proteins from the dynamin family, which polymerize into helical spirals around such necks. Much research has been devoted to elucidating the specifics of that “somehow”, but despite no shortage of ideas, the question is not settled. Pictorially obvious notions of “strangling” or “pushing” are difficult to render in mechanically precise terms. Moreover, since dynamin is a GTPase, it is tempting to speculate that it has a motor activity that assists the necessary severing “action”, but again the underlying mechanics is not obvious. We believe the difficulty to be the mechanically nontrivial nature of confining elastic filaments onto curved surfaces, for which efficient methods to conceptualize the associated forces and torques have only recently appeared. Here we investigate the implications of a conceptually simple yet mechanically challenging model: consider an elastic helical filament confined to a surface mimicking the neck between two membrane compartments, which we assume to take the shape of a catenoid. What can we say about the expected length of such adsorbed filaments, their shapes, and the forces they exert, as a function of the key parameters in the model? While real dynamin is surely more complex, we consider such a minimal model to be the indispensable baseline. Without knowing what such a model can and cannot explain, it is difficult to justify more complex mechanisms, or understand the constraints under which this machinery evolved in the first place.
|
|
Modern simulation approaches in soft matter science:
From fundamental understanding to industrial applications
Festschrift on the occasion of Kurt Kremer’s 60th birthday
Luigi Delle Site, Markus Deserno, Burkhard Dünweg, Christian Holm, Christine Peter, and Harald Pleiner
Eur. Phys. J. Sp. Topics 225, 1317–1321 (2016).
 This special topics issue offers a broad perspective on recent theoretical and computational soft matter science, providing state of the art advances in many of its sub-fields. As is befitting for a discipline as diverse as soft matter, the papers collected here span a considerable range of subjects and questions, but they also illustrate numerous connections into both fundamental science and technological/industrial applications, which have accompanied the field since its earliest days. This issue is dedicated to Kurt Kremer, on the occasion of his 60th birthday, honouring his role in establishing this exciting field and consolidating its standing in the frame of current science and technology. This special topics issue offers a broad perspective on recent theoretical and computational soft matter science, providing state of the art advances in many of its sub-fields. As is befitting for a discipline as diverse as soft matter, the papers collected here span a considerable range of subjects and questions, but they also illustrate numerous connections into both fundamental science and technological/industrial applications, which have accompanied the field since its earliest days. This issue is dedicated to Kurt Kremer, on the occasion of his 60th birthday, honouring his role in establishing this exciting field and consolidating its standing in the frame of current science and technology.
A link to the whole special issue is can be found under http://link.springer.com/journal/11734/225/8 |
|
Effect of intrinsic curvature and edge tension on the stability of binary mixed-membrane three-junctions
Jasmine M. Gardner, Markus Deserno, and Cameron F. Abrams
J. Chem. Phys. 145, 074901 (2016). (Feature article)
 We use a combination of coarse-grained molecular dynamics simulations and theoretical modeling to examine three-junctions in mixed lipid bilayer membranes. These junctions are localized defect lines in which three bilayers merge in such a way that each bilayer shares one monolayer with one of the other two bilayers. The resulting local morphology is non-lamellar, resembling the three-fold symmetric defect lines in inverse hexagonal phases, but it regularly occurs during membrane fission and fusion events. We realize a system of junctions by setting up a honeycomb lattice, which in its primitive cell contains two hexagons and four three-line junctions, permitting us to study their stability as well as their line tension. We specifically consider the effects of lipid composition and intrinsic curvature in binary mixtures, which contain a fraction of negatively curved lipids in a curvature-neutral background phase. Three-junction stability results from a competition between the junction and an open edge, which arises if one of the three bilayers detaches from the other two. We show that the stable phase is the one with the lower defect line tension. The strong and opposite monolayer curvatures present in junctions and edges enhance the mole fraction of negatively curved lipids in junctions and deplete it in edges. This lipid sorting affects the two line tensions and in turn the relative stability of the two phases. It also leads to a subtle entropic barrier for the transition between junction and edge that is absent in uniform membranes. We use a combination of coarse-grained molecular dynamics simulations and theoretical modeling to examine three-junctions in mixed lipid bilayer membranes. These junctions are localized defect lines in which three bilayers merge in such a way that each bilayer shares one monolayer with one of the other two bilayers. The resulting local morphology is non-lamellar, resembling the three-fold symmetric defect lines in inverse hexagonal phases, but it regularly occurs during membrane fission and fusion events. We realize a system of junctions by setting up a honeycomb lattice, which in its primitive cell contains two hexagons and four three-line junctions, permitting us to study their stability as well as their line tension. We specifically consider the effects of lipid composition and intrinsic curvature in binary mixtures, which contain a fraction of negatively curved lipids in a curvature-neutral background phase. Three-junction stability results from a competition between the junction and an open edge, which arises if one of the three bilayers detaches from the other two. We show that the stable phase is the one with the lower defect line tension. The strong and opposite monolayer curvatures present in junctions and edges enhance the mole fraction of negatively curved lipids in junctions and deplete it in edges. This lipid sorting affects the two line tensions and in turn the relative stability of the two phases. It also leads to a subtle entropic barrier for the transition between junction and edge that is absent in uniform membranes.
|
|
Breaking a Virus: Identifying Molecular Level Failure Modes of a Viral Capsid by Multiscale Modeling
Venkatramanan Krishnamani, Christoph Globisch, Christine Peter, and Markus Deserno
Eur. Phys. J. Sp. Topics 225, 1757–1774 (2016).
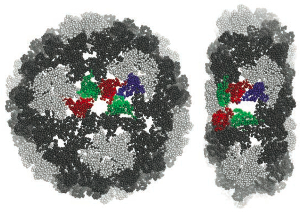 We use coarse-grained (CG) simulations to study the de- formation of empty Cowpea Chlorotic Mottle Virus (CCMV) capsids under uniaxial compression, from the initial elastic response up to capsid breakage. Our CG model is based on the MARTINI force field and has been amended by a stabilizing elastic network, acting only within individual proteins, that was tuned to capture the fluctuation spectrum of capsid proteins dimers, obtained from all atom simulations. We have previously shown that this model predicts force-compression curves that match AFM indentation experiments on empty CCMV capsids. Here we investigate details of the actual breaking events in CCMV capsid during mechanical compression. We present a symmetry classification of all relevant protein contacts and show that they differ significantly in terms of stability. Specifically, we show that interfaces which break readily are precisely those which are believed to form last during assembly, even though some of them might share virtually the same contacts as other non-breaking interfaces. Specifically, the interfaces that form pentamers of dimers never break, while the virtually identical interface between hexamers of dimers readily do so. Since these units differ in the large-scale geometry and, in particular, the cone-angle at the center of the 5- or 6-fold vertex, we propose that the hexametric unit fails because it is pre-stressed. This not only suggests that hexamers of dimers form less frequently during the early stages of assembly; it also offers a natural explanation for the well-known β-barrel motif at the hexametric center as a post-aggregation stabilization mechanism. Finally, we identify those amino acid contacts within all key protein interfaces that are most persistent during com- pressive deformation of the capsid, thereby providing potential targets for mutation studies aiming to elucidate the key contacts upon which overall stability rests. We use coarse-grained (CG) simulations to study the de- formation of empty Cowpea Chlorotic Mottle Virus (CCMV) capsids under uniaxial compression, from the initial elastic response up to capsid breakage. Our CG model is based on the MARTINI force field and has been amended by a stabilizing elastic network, acting only within individual proteins, that was tuned to capture the fluctuation spectrum of capsid proteins dimers, obtained from all atom simulations. We have previously shown that this model predicts force-compression curves that match AFM indentation experiments on empty CCMV capsids. Here we investigate details of the actual breaking events in CCMV capsid during mechanical compression. We present a symmetry classification of all relevant protein contacts and show that they differ significantly in terms of stability. Specifically, we show that interfaces which break readily are precisely those which are believed to form last during assembly, even though some of them might share virtually the same contacts as other non-breaking interfaces. Specifically, the interfaces that form pentamers of dimers never break, while the virtually identical interface between hexamers of dimers readily do so. Since these units differ in the large-scale geometry and, in particular, the cone-angle at the center of the 5- or 6-fold vertex, we propose that the hexametric unit fails because it is pre-stressed. This not only suggests that hexamers of dimers form less frequently during the early stages of assembly; it also offers a natural explanation for the well-known β-barrel motif at the hexametric center as a post-aggregation stabilization mechanism. Finally, we identify those amino acid contacts within all key protein interfaces that are most persistent during com- pressive deformation of the capsid, thereby providing potential targets for mutation studies aiming to elucidate the key contacts upon which overall stability rests.
|
|
Determining the Lipid Tilt Modulus by Simulating Membrane Buckles
Xin Wang (王昕) and Markus Deserno
J. Phys. Chem. B 120, 6061–6073 (2016).
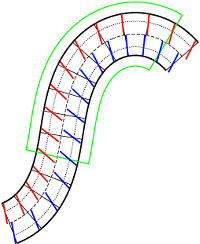 Lipid tilt affects the energy of membrane deformations on scales comparable to the membrane's thickness. The surface divergence of the tilt field creates a local spontaneous curvature, while tilt itself is quadratically penalized with a strength given by the tilt modulus. Traditionally, this modulus is determined by measuring the power spectrum of lipid orientation fluctuations. Here we present a novel approach which does not rely on fluctuations but instead exploits the fact that curvature gradients induce a tilt field. Its implementation extends a technique previously developed by us for localizing the position of the pivotal plane in buckling simulations, which quantifies the lipid imbalance across segments cut out from a complete buckle. Lipid tilt affects this count in a predictable way, and the signal can be quantified well enough to back out the tilt modulus—at no additional cost and with about 5% precision for not too coarse models. We apply our technique to three lipid models of very different resolution: the highly coarse grained Cooke model, and two versions of DMPC, using both the (less highly coarse grained) MARTINI and the (united atom) Berger force field. For Cooke we find an effective bilayer tilt modulus of 29±9 pN/nm, and for the less generic DMPC lipid we find 115±6 pN/nm for MARTINI and 39±2 pN/nm for Berger, both in reasonable agreement with existing studies for these models. We also show that the position of the pivotal plane for Berger DMPC lies just below the glycerol backbone—unlike for MARTINI DMPC, where this plane lies closer to the middle of the lipid. Lipid tilt affects the energy of membrane deformations on scales comparable to the membrane's thickness. The surface divergence of the tilt field creates a local spontaneous curvature, while tilt itself is quadratically penalized with a strength given by the tilt modulus. Traditionally, this modulus is determined by measuring the power spectrum of lipid orientation fluctuations. Here we present a novel approach which does not rely on fluctuations but instead exploits the fact that curvature gradients induce a tilt field. Its implementation extends a technique previously developed by us for localizing the position of the pivotal plane in buckling simulations, which quantifies the lipid imbalance across segments cut out from a complete buckle. Lipid tilt affects this count in a predictable way, and the signal can be quantified well enough to back out the tilt modulus—at no additional cost and with about 5% precision for not too coarse models. We apply our technique to three lipid models of very different resolution: the highly coarse grained Cooke model, and two versions of DMPC, using both the (less highly coarse grained) MARTINI and the (united atom) Berger force field. For Cooke we find an effective bilayer tilt modulus of 29±9 pN/nm, and for the less generic DMPC lipid we find 115±6 pN/nm for MARTINI and 39±2 pN/nm for Berger, both in reasonable agreement with existing studies for these models. We also show that the position of the pivotal plane for Berger DMPC lies just below the glycerol backbone—unlike for MARTINI DMPC, where this plane lies closer to the middle of the lipid.
|
|
2015 |
| |
Folding and insertion thermodynamics of the transmembrane WALP peptide
Tristan Bereau, W. F. Drew Bennett, Jim Pfaendtner, Markus Deserno, and Mikko Karttunen
J. Chem. Phys. 143, 243127 (2015).
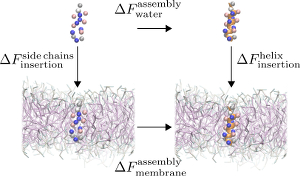 The anchor of most integral membrane proteins consists of one or several helices spanning the lipid bilayer. The WALP peptide, GWW(LA)n(L)WWA, is a common model helix to study the fundamentals of protein insertion and folding, as well as helix-helix association in the membrane. Its structural properties have been illuminated in a large number of experimental and simulation studies. In this combined coarse-grained and atomistic simulation study, we probe the thermodynamics of a single WALP peptide, focusing on both the insertion across the water-membrane interface, as well as folding in both water and a membrane. The potential of mean force characterizing the peptide's insertion into the membrane shows qualitatively similar behavior across peptides and three force fields. However, the Martini force field exhibits a pronounced secondary minimum for an adsorbed interfacial state, which may even become the global minimum—in contrast to both atomistic simulations and the alternative PLUM force field. Even though the two coarse-grained models reproduce the free energy of insertion of individual amino acids side chains, they both underestimate its corresponding valu for the full peptide (as compared with atomistic simulations), hinting at cooperative physics beyond the residue level. Folding of WALP in the two environments indicates the helix as the most stable structure, though with different relative stabilities and chain-length dependence. The anchor of most integral membrane proteins consists of one or several helices spanning the lipid bilayer. The WALP peptide, GWW(LA)n(L)WWA, is a common model helix to study the fundamentals of protein insertion and folding, as well as helix-helix association in the membrane. Its structural properties have been illuminated in a large number of experimental and simulation studies. In this combined coarse-grained and atomistic simulation study, we probe the thermodynamics of a single WALP peptide, focusing on both the insertion across the water-membrane interface, as well as folding in both water and a membrane. The potential of mean force characterizing the peptide's insertion into the membrane shows qualitatively similar behavior across peptides and three force fields. However, the Martini force field exhibits a pronounced secondary minimum for an adsorbed interfacial state, which may even become the global minimum—in contrast to both atomistic simulations and the alternative PLUM force field. Even though the two coarse-grained models reproduce the free energy of insertion of individual amino acids side chains, they both underestimate its corresponding valu for the full peptide (as compared with atomistic simulations), hinting at cooperative physics beyond the residue level. Folding of WALP in the two environments indicates the helix as the most stable structure, though with different relative stabilities and chain-length dependence.
|
|
Determining the pivotal plane of fluid lipid membranes in simulations
Xin Wang (王昕) and Markus Deserno
J. Chem. Phys. 143, 164109 (2015).
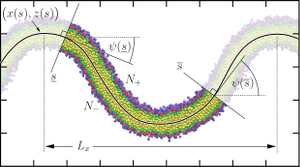 Each leaflet of a curved lipid membrane contains a surface at which the area strain vanishes, the so-called pivotal plane. Its distance z0 from the bilayer's midplane arises in numerous contexts, for instance the connection between monolayer and bilayer moduli, stress-profile moments, or area-difference elasticity theories. Here we propose two precise methods for determining the location of the pivotal plane in computer simulations, both of which rely on monitoring the lipid imbalance across a curved bilayer. The first method considers the ratio of lipid number between the two leaflets of cylindrical or spherical vesicles; it hence requires lipid flip-flop for equilibration. The second method looks at the leaflet difference across local sections cut out from a buckled membrane; this observable equilibrates even in the absence of flip-flop. We apply our methods to two different coarse-grained lipid models, the generic three-bead solvent-free Cooke model, and a ten-bead representation of dimyristoylphosphocholine (DMPC) with the explicit solvent MARTINI model. The Cooke model is amenable to both methods and gives results that agree at the percent level. Using it, we also show that the pivotal plane moves outward as lipid curvature becomes more positive. The MARTINI model can only be analyzed with the buckling method; the obtained value z0=0.850(11)nm lies about 0.4nm inwards of the glycerol backbone and is hence unexpectedly small. We attribute this to limitations of the coarse-grained description, suggesting that the location of the pivotal plane might be a good indicator for how well lipid models capture the microscopic origins of curvature elasticity. Finally, we also show that the pivotal plane position itself moves as the membrane is bent. The leading correction is linear in curvature, dependent on the Poisson ratio, and can matter when analyzing experimental results obtained from highly curved inverse hexagonal phases. Each leaflet of a curved lipid membrane contains a surface at which the area strain vanishes, the so-called pivotal plane. Its distance z0 from the bilayer's midplane arises in numerous contexts, for instance the connection between monolayer and bilayer moduli, stress-profile moments, or area-difference elasticity theories. Here we propose two precise methods for determining the location of the pivotal plane in computer simulations, both of which rely on monitoring the lipid imbalance across a curved bilayer. The first method considers the ratio of lipid number between the two leaflets of cylindrical or spherical vesicles; it hence requires lipid flip-flop for equilibration. The second method looks at the leaflet difference across local sections cut out from a buckled membrane; this observable equilibrates even in the absence of flip-flop. We apply our methods to two different coarse-grained lipid models, the generic three-bead solvent-free Cooke model, and a ten-bead representation of dimyristoylphosphocholine (DMPC) with the explicit solvent MARTINI model. The Cooke model is amenable to both methods and gives results that agree at the percent level. Using it, we also show that the pivotal plane moves outward as lipid curvature becomes more positive. The MARTINI model can only be analyzed with the buckling method; the obtained value z0=0.850(11)nm lies about 0.4nm inwards of the glycerol backbone and is hence unexpectedly small. We attribute this to limitations of the coarse-grained description, suggesting that the location of the pivotal plane might be a good indicator for how well lipid models capture the microscopic origins of curvature elasticity. Finally, we also show that the pivotal plane position itself moves as the membrane is bent. The leading correction is linear in curvature, dependent on the Poisson ratio, and can matter when analyzing experimental results obtained from highly curved inverse hexagonal phases.
|
|
Curvature softening and negative compressibility of gel-phase lipid membranes
Patrick Diggins IV, Zachary A. McDargh, and Markus Deserno
J. Am. Chem. Soc. 137, 12752–12755 (2015).
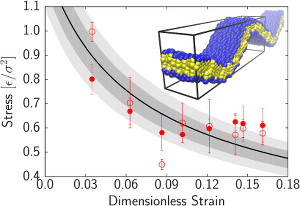 We show that gel-phase lipid membranes soften upon bending, leading to curvature localization and a negative compressibility. Using simulations of two very different lipid models to quantify shape and stress-strain relation of buckled membranes, we demonstrate that gel phase bilayers do not behave like Euler elastica and hence are not well described by a quadratic Helfrich Hamiltonian—much unlike their fluid-phase counterparts. We propose a theoretical framework which accounts for the observed softening through an energy density that smoothly crosses over from a quadratic to a linear curvature dependence beyond a critical new scale ℓ-1. This simple model captures both the shape and the stress-strain relation for our two sets of simulations and permits the extraction of bending moduli, which are found to be about an order of magnitude larger than the corresponding fluid phase values. We also find surprisingly large cross-over lengths ℓ, several times bigger than the bilayer thickness, rendering the exotic elasticity of gel-phase membranes more strongly pronounced than that of homogeneous compressible sheets and even artificial metamaterials. We therefore suggest that such membranes have unexpected potential as nano-scale systems with striking materials characteristics. We show that gel-phase lipid membranes soften upon bending, leading to curvature localization and a negative compressibility. Using simulations of two very different lipid models to quantify shape and stress-strain relation of buckled membranes, we demonstrate that gel phase bilayers do not behave like Euler elastica and hence are not well described by a quadratic Helfrich Hamiltonian—much unlike their fluid-phase counterparts. We propose a theoretical framework which accounts for the observed softening through an energy density that smoothly crosses over from a quadratic to a linear curvature dependence beyond a critical new scale ℓ-1. This simple model captures both the shape and the stress-strain relation for our two sets of simulations and permits the extraction of bending moduli, which are found to be about an order of magnitude larger than the corresponding fluid phase values. We also find surprisingly large cross-over lengths ℓ, several times bigger than the bilayer thickness, rendering the exotic elasticity of gel-phase membranes more strongly pronounced than that of homogeneous compressible sheets and even artificial metamaterials. We therefore suggest that such membranes have unexpected potential as nano-scale systems with striking materials characteristics.
|
|
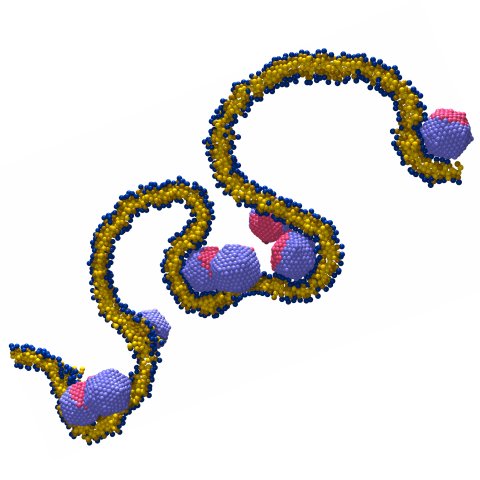
Design Principles for Nanoparticles Enveloped by a Polymer-Tethered Lipid Membrane
Mingyang Hu, Francesca Stanzione, Amadeu K. Sum, Roland Faller, and Markus Deserno
ACS Nano 9, 9942–9954 (2015).
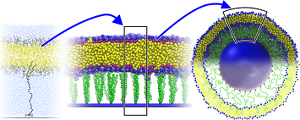 We propose the design for a nanoparticle carrier that combines three existing motifs into a single construct: a liposome is stabilized by anchoring it to an enclosed solid core via extended polymeric tethers that are chemically grafted to the core and physisorb into the surrounding lipid membrane. Such a design would exhibit several enticing properties, among them: (i) the anchoring stabilizes the liposome against a variety of external stresses, while preserving an aqueous compartment between core and membrane; (ii) the interplay of design parameters such as polymer length or grafting density enforces strong constraints on nanoparticle size and hence ensures a high degree of uniformity; and (iii) the physical and chemical characteristics of the individual constituents equip the construct with numerous functionalities that can be exploited in many ways. However, navigating the large parameter space requires a sound prior understanding for how various design features work together, and how this impacts potential pathways for synthesizing and assembling these nanoparticles. In this paper we examine these connections in detail, using both soft matter theory and computer simulations at all levels of resolution. We thereby derive strong constraints on the experimentally relevant parameter space, and also propose potential equilibrium and non-equilibrium pathways for nanoparticle assembly. We propose the design for a nanoparticle carrier that combines three existing motifs into a single construct: a liposome is stabilized by anchoring it to an enclosed solid core via extended polymeric tethers that are chemically grafted to the core and physisorb into the surrounding lipid membrane. Such a design would exhibit several enticing properties, among them: (i) the anchoring stabilizes the liposome against a variety of external stresses, while preserving an aqueous compartment between core and membrane; (ii) the interplay of design parameters such as polymer length or grafting density enforces strong constraints on nanoparticle size and hence ensures a high degree of uniformity; and (iii) the physical and chemical characteristics of the individual constituents equip the construct with numerous functionalities that can be exploited in many ways. However, navigating the large parameter space requires a sound prior understanding for how various design features work together, and how this impacts potential pathways for synthesizing and assembling these nanoparticles. In this paper we examine these connections in detail, using both soft matter theory and computer simulations at all levels of resolution. We thereby derive strong constraints on the experimentally relevant parameter space, and also propose potential equilibrium and non-equilibrium pathways for nanoparticle assembly.
|
|
Cylindrical confinement of semiflexible polymers
Pablo Vázquez-Montejo, Zachary McDargh, Markus Deserno, and Jemal Guven
Phys. Rev. E 91, 063203 (2015).
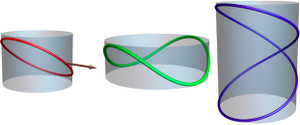 Equilibrium states of a closed semiflexible polymer binding to a cylinder are described. This may be either by confinement or by constriction. Closed completely bound states are labeled by two integers: the number of oscillations, n, and the number of times it winds the cylinder, p, the latter being a topological invariant. We examine the behavior of these states as the length of the loop is increased by evaluating the energy, the conserved axial torque, and the contact force. The ground state for a given p is the state with n = 1; a short loop with p = 1 is an elliptic deformation of a parallel circle; as its length increases it elongates along the cylinder axis with two hairpin ends. Excited states with n ≥ 2 and p = 1 possess n-fold axial symmetry. Short (long) loops possess energies E ≈ pE0 (nE0), with E0 the energy of a circular loop with same radius as the cylinder; in long loops the axial torque vanishes. Confined bound excited states are initially unstable; however, above a critical length each n-fold state becomes stable: The folded hairpin cannot be unfolded. The ground state for each p is also initially unstable with respect to deformations rotating the loop off the surface into the interior. A closed planar elastic curve aligned along the cylinder axis making contact with the cylinder on its two sides is identified as the ground state of a confined loop. Exterior bound states behave very differently, if free to unbind, as signaled by the reversal in the sign of the contact force. If p = 1, all such states are unstable. If p ≥ 2, however, a topological obstruction to complete unbinding exists. If the loop is short, the bound state with p = 2 and n = 1 provides a stable constriction of the cylinder, partially unbinding as the length is increased. This motif could be relevant to an understanding of the process of membrane fission mediated by dynamin rings. Equilibrium states of a closed semiflexible polymer binding to a cylinder are described. This may be either by confinement or by constriction. Closed completely bound states are labeled by two integers: the number of oscillations, n, and the number of times it winds the cylinder, p, the latter being a topological invariant. We examine the behavior of these states as the length of the loop is increased by evaluating the energy, the conserved axial torque, and the contact force. The ground state for a given p is the state with n = 1; a short loop with p = 1 is an elliptic deformation of a parallel circle; as its length increases it elongates along the cylinder axis with two hairpin ends. Excited states with n ≥ 2 and p = 1 possess n-fold axial symmetry. Short (long) loops possess energies E ≈ pE0 (nE0), with E0 the energy of a circular loop with same radius as the cylinder; in long loops the axial torque vanishes. Confined bound excited states are initially unstable; however, above a critical length each n-fold state becomes stable: The folded hairpin cannot be unfolded. The ground state for each p is also initially unstable with respect to deformations rotating the loop off the surface into the interior. A closed planar elastic curve aligned along the cylinder axis making contact with the cylinder on its two sides is identified as the ground state of a confined loop. Exterior bound states behave very differently, if free to unbind, as signaled by the reversal in the sign of the contact force. If p = 1, all such states are unstable. If p ≥ 2, however, a topological obstruction to complete unbinding exists. If the loop is short, the bound state with p = 2 and n = 1 provides a stable constriction of the cylinder, partially unbinding as the length is increased. This motif could be relevant to an understanding of the process of membrane fission mediated by dynamin rings.
|
|
Fluid lipid membranes: From differential geometry to curvature stresses
Markus Deserno
Chem. Phys. Lipids, 185, 11–45 (2015).
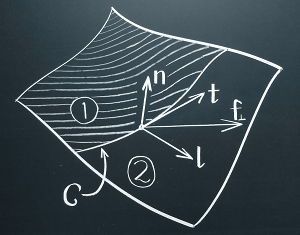
A fluid lipid membrane transmits stresses and torques that are fully determined by its geometry. They can be described by a stress- and torque-tensor, respectively, which yield the force or torque per length through any curve drawn on the membrane's surface. In the absence of external forces or torques the surface divergence of these tensors vanishes, revealing them as conserved quantities of the underlying Euler-Lagrange equation for the membrane's shape. This review provides a comprehensive introduction into these concepts without assuming the reader's familiarity with differential geometry, which instead will be developed as needed, relying on little more than vector calculus. The Helfrich Hamiltonian is then introduced and discussed in some depth. By expressing the quest for the energy-minimizing shape as a functional variation problem subject to geometric constraints, as proposed by Guven [J. Phys. A: Math. Gen. 37, L313–9 (2004)], stress and torque tensors naturally emerge, and their connection to the shape equation becomes evident. How to reason with both tensors is then illustrated with a number of simple examples, after which this review concludes with four more sophisticated applications: boundary conditions for adhering membranes, corrections to the classical micropipette aspiration equation, membrane buckling, and membrane mediated interactions. |
|
Enhanced Sampling of Coarse-Grained Transmembrane-Peptide Structure Formation from Hydrogen-Bond Replica Exchange
Tristan Bereau and Markus Deserno
J. Membrane Biol. 248, 395–405 (2015).
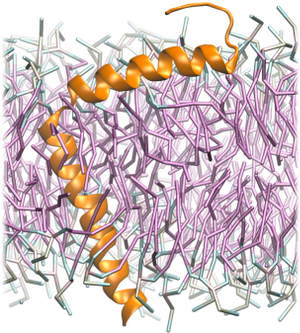
Protein structure formation in the membrane highlights a grand challenge of sampling in computer simulations, because kinetic traps and slow dynamics make it difficult to find the native state. Exploiting increased fluctuations at higher temperatures can help overcome free-energy barriers, provided the membrane's structure remains stable. In this work, we apply Hamiltonian replica-exchange molecular dynamics, where we only tune the backbone hydrogen-bond strength to help reduce the propensity of long-lived misfolded states. Using a recently developed coarse-grained model, we illustrate the robustness of the method by folding different WALP transmembrane helical peptides starting from stretched, unstructured conformations. We show the efficiency of the method by comparing to simulations without enhanced sampling, achieving folding in one example after significantly longer simulation times. Analysis of the bilayer structure during folding provides insight into the local membrane deformation during helix formation as a function of chain length (from 16 to 23 residues). Finally, we apply our method to fold the 50-residue-long major pVIII coat protein (fd coat) of the filamentous fd bacteriophage. Our results agree well with experimental structures and atomistic simulations based on implicit membrane models, suggesting that our explicit CG folding protocol can serve as a starting point for better-refined atomistic simulations in a multiscale framework. |
|
2014 |
| |
An effective field theory of thermal Casimir interactions between anisotropic particles
Robert C. Haussman and Markus Deserno
Phys. Rev. E 89, 062102 (2014).
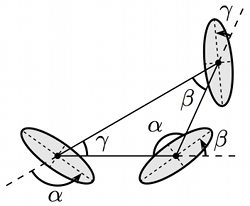
We employ an effective field theory (EFT) approach to study thermal Casimir interactions between objects bound to a fluctuating fluid surface or interface dominated by surface tension, with a focus on the effects of anisotropic particles. The EFT prescription disentangles the constraints imposed by the particles' boundaries from the calculation of the interaction free energy by constructing an equivalent point particle description. The finite-size information is captured in a derivative expansion that encodes the particles' response to external fields. The coefficients of the expansion terms correspond to generalized polarizabilities and are found by matching the results of a linear response boundary value problem computed in both the full and effective theories. We demonstrate the versatility of the EFT approach by constructing the general effective Hamiltonian for a collection of particles of arbitrary shapes. Taking advantage of the conformal symmetry of the Hamiltonian, we discuss a straightforward conformal mapping procedure to systematically determine the polarizabilities and derive a complete description for elliptical particles. We compute the pairwise interaction energies to several orders for nonidentical ellipses as well as their leading-order triplet interactions. The resulting preferred pair and multibody configurations are discussed. |
|
More than the Sum of its Parts: Coarse-Grained Peptide-Lipid Interactions from a Simple Cross-Parametrization
Tristan Bereau, Zun-Jing Wang, and Markus Deserno
J. Chem. Phys. 140, 115101 (2014). (Feature article)

Interfacial systems are at the core of fascinating phenomena in many disciplines, such as biochemistry, soft-matter physics, and food science. However, the parametrization of accurate, reliable, and consistent coarse-grained (CG) models for systems at interfaces remains a challenging endeavor. In the present work we explore to what extent two independently developed solvent-free CG models of peptides and lipids—of different mapping schemes, parametrization methods, target functions, and validation criteria—can be combined by only tuning the cross interactions. Our results show that the cross-parametrization can reproduce a number of structural properties of membrane peptides (for example tilt and hydrophobic mismatch), in agreement with existing peptide-lipid CG force fields. We find encouraging results for two challenging biophysical problems: (i) membrane pore formation mediated by the cooperative action of several antimicrobial peptides, and (ii) the insertion and folding of the helix-forming peptide WALP23 in the membrane. |
|
The Effective Field Theory approach towards\\ membrane-mediated interactions between particles
Cem Yolcu, Robert C. Haussman, and Markus Deserno
Adv. Colloid Interface Sci. 208, 89–109 (2014).
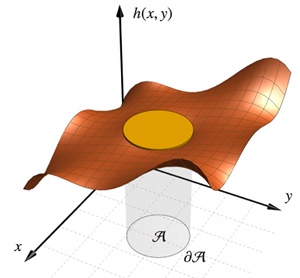
Fluid lipid membranes can mediate forces between particles bound to them: A local deformation of the surface geometry created by some object spreads to distant regions, where other objects can respond to it. The physical characteristics of these geometric interactions, and how they are affected by thermal fluctuations, are well described by the simple continuum curvature-elastic Hamiltonian proposed 40 years ago by Wolfgang Helfrich. Unfortunately, while the underlying principles are conceptually straightforward, the corresponding calculations are not—largely because one must enforce boundary conditions for finite-sized objects. This challenge has inspired several heuristic approaches for expressing the problem in a point particle language. While streamlining the calculations of leading order results and enabling predictions for higher order corrections, the ad hoc nature of the reformulation leaves its domain of validity unclear. In contrast, the framework of Effective Field Theory (EFT) provides a systematic way to construct a completely equivalent point particle description. In this review we present a detailed account for how this is accomplished. In particular, we use a familiar example from electrostatics as an analogy to motivate the key steps needed to construct an EFT, most notably capturing finite size information in point-like "polarizabilities," and determining their value through a suitable "matching procedure." The interaction (free) energy then emerges as a systematic cumulant expansion, for which powerful diagrammatic techniques exist, which we also briefly revisit. We then apply this formalism to derive series expansions for interactions between flat and curved particle pairs, multibody interactions, as well as corrections to all these interactions due to thermal fluctuations. |
|
2013 |
| |
Computational Studies of Biomembrane Systems: Theoretical Considerations, Simulation Models, and Applications
Markus Deserno, Kurt Kremer, Harald Paulsen, Christine Peter, and Friederike Schmid
Adv. Polym. Sci., 1–47 (2013).
This chapter summarizes several approaches combining theory, simula- tion, and experiment that aim for a better understanding of phenomena in lipid bilayers and membrane protein systems, covering topics such as lipid rafts, membrane-mediated interactions, attraction between transmembrane proteins, and aggregation in biomembranes leading to large superstructures such as the light- harvesting complex of green plants. After a general overview of theoretical con- siderations and continuum theory of lipid membranes we introduce different options for simulations of biomembrane systems, addressing questions such as: What can be learned from generic models? When is it expedient to go beyond them? And, what are the merits and challenges for systematic coarse graining and quasi-atomistic coarse-grained models that ensure a certain chemical specificity? |
|
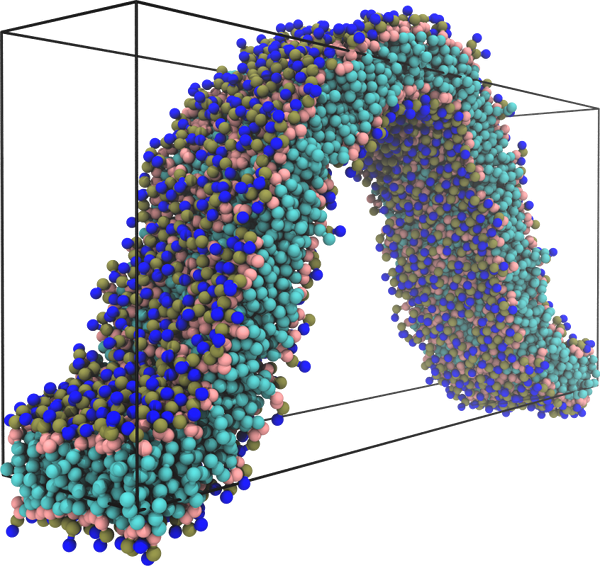
Determining the bending modulus of a lipid membrane by simulating buckling
Mingyang Hu (胡明暘), Patrick Diggins IV, and Markus Deserno
J. Chem. Phys. 138, 214110 (2013).
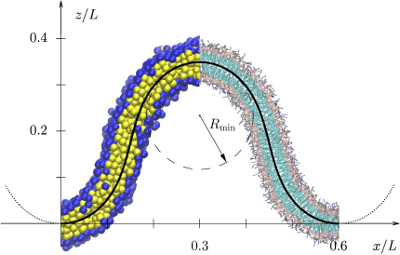
The force needed to buckle a thin elastic surface is proportional to its bending rigidity. This fact suggests using a buckling setup to measure the bending modulus of lipid membranes. Extending the work of Noguchi [Phys. Rev. E 83, 061919 (2011)], we systematically derive highly accurate analytical expressions for the forces along and perpendicular to the buckle, and we elucidate some of their counterintuitive properties using the framework of a surface stress tensor. Furthermore, we estimate the corrections to buckling forces due to thermal fluctuations and find them significant only for stresses along the ridges. We then apply this buckling protocol to four different lipid membrane models, which widely differ in their level of resolution and the treatment of solvent, and show that in all cases buckling is a reliable and accurate means for measuring their rigidity. Finally, we show that monitoring both stresses and energies during a simulation offers additional insights into the thermodynamics of curvature elasticity and permits one to predict the bending rigidity for a range of temperatures around the actual simulation temperature. |
|
Optimization of an Elastic Network Augmented Coarse Grained Model to Study CCMV Capsid Deformation
Christoph Globisch, Venkatramanan Krishnamani, Markus Deserno, and Christine Peter
PLOS ONE, 8(4): e60582. doi:10.1371/journal.pone.0060582.
The major protective coat of most viruses is a highly symmetric protein capsid that forms spontaneously from many copies of identical proteins. Structural and mechanical properties of such capsids, as well as their self-assembly process, have been studied experimentally and theoretically, including modeling efforts by computer simulations on various scales. Atomistic models include specific details of local protein binding but are limited in system size and accessible time, while coarse grained (CG) models do get access to longer time and length scales but often lack the specific local interactions. Multi-scale models aim at bridging this gap by systematically connecting different levels of resolution. Here, a CG model for CCMV (Cowpea Chlorotic Mottle Virus), a virus with an icosahedral shell of 180 identical protein monomers, is developed, where parameters are derived from atomistic simulations of capsid protein dimers in aqueous solution. In particular, a new method is introduced to combine the MARTINI CG model with a supportive elastic network based on structural fluctuations of individual monomers. In the parametrization process, both network connectivity and strength are optimized. This elastic-network optimized CG model, which solely relies on atomistic data of small units (dimers), is able to correctly predict inter-protein conformational flexibility and properties of larger capsid fragments of 20 and more subunits. Furthermore, it is shown that this CG model reproduces experimental (Atomic Force Microscopy) indentation measurements of the entire viral capsid. Thus it is shown that one obvious goal for hierarchical modeling, namely predicting mechanical properties of larger protein complexes from models that are carefully parametrized on elastic properties of smaller units, is achievable. |
|
Gaussian curvature elasticity determined from global shape transformations and local stress distributions: a comparative study using the MARTINI model
Mingyang Hu, Djurre H. de Jong, Siewert J. Marrink, and Markus Deserno
Faraday Discuss. 161, 365–382 (2013).
We calculate the Gaussian curvature modulus
κ of a systematically coarsegrained (CG) one-component lipid membrane by applying the method recently proposed by Hu et al. [Biophys. J., 2012, 102, 1403] to the MARTINI representation of 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC). We find the value
κ/κ= –1.04∓0.03 for the elastic ratio between the Gaussian and the mean curvature modulus and deduce
κm/κm= –0.95∓0.09 for the monolayer elastic ratio, where the latter is based on plausible assumptions for the distance z0 of the monolayer neutral surface from the bilayer midplane and the spontaneous lipid curvature K0m. By also analyzing the lateral stress profile σ0(z) of our system, two other lipid types and pertinent data from the literature, we show that determining K0m and
κ through the first and second moment of σ0(z) gives rise to physically implausible values for these observables. This discrepancy, which we previously observed for a much simpler CG model, suggests that the moment conditions derived from simple continuum assumptions miss the effect of physically important correlations in the lipid bilayer.
|
|
2012 |
| |
Membrane-mediated interactions between rigid inclusions: An effective field theory
Cem Yolcu and Markus Deserno
Phys. Rev. E 86, 031906 (2012).
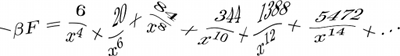
An approach based on effective field theory (EFT) is discussed and applied to the problem of surface-mediated interactions between rigid inclusions of circular footprint on a membrane. Instead of explicitly constraining the surface fluctuations in accord with the boundary conditions around the inclusions, the EFT formalism rewrites the theory; the Hamiltonian of a freely fluctuating surface is augmented by pointwise localized terms that capture the same constraints. This allows one to compute the interaction free energy as an asymptotic expansion in inverse separations in a systematic, efficient and transparent way. Both entropic (fluctuation-induced, Casimir-like) and curvature-elastic (ground-state) forces are considered. Our findings include higher order corrections to known asymptotic results, both on the pair and multibody level. We also found that the few previous at- tempts in the literature at predicting subleading orders missed some terms due to an uncontrolled point-particle approximation. |
|
Hemifusion of giant unilamellar vesicles with planar hydrophobic surfaces:
A fluorescence microscopy study
Goh Haw Zan, Cheemeng Tan, Markus Deserno, Frederick Lanni, and Mathias Lösche
Soft Matter 8, 10877–10886 (2012).

Vesicle adhesion and fusion to interfaces are frequently used for the construction of biomimetic surfaces in biosensors and drug delivery. Ubiquitous in cell biology, vesicle fusion involves the transformation of two separate membranes into one contiguous lipid bilayer. In distinction, the deposition of vesicle membranes to hydrophobic surfaces requires the transformation of a lipidic bilayer into a monomolecular layer—a topologically distinct process termed hemifusion. Here, we used hydrophobically terminated self-assembled monolayers (SAM) on solid surfaces to track the fusion of fluorescently labeled giant unilamellar vesicles (GUVs) at the single vesicle level with video time resolution (≈ 53 ms). We observed that a dilute monolayer, consisting of lipid abstracted from the outer GUV leaflet, spreads outward across the hydrophobic surface from the vesicle adhesion site. Subsequently, bilayer fusion occurs by vesicle rupture near the hydrophobic surface, followed by spreading of lipid in a dense monolayer. GUV lipids thus transfer to the SAM surface in two concentric zones: an outer fusion zone comprises lipids drawn from the outer GUV leaflet; an inner fusion zone comprises lipids from both the inner and outer GUV leaflets and grows at a rate of ≈ 1000 μm2/s (dA/dt = 970 ± 430 μm2/s in n = 22 independent experiments). This growth rate is quantitatively consistent with the assumption that the spreading of the monolayer is entirely driven by the difference in surface energies of the hydrophobic and the lipid-covered SAM surfaces, which is dissipated by friction of the spreading monolayer on the SAM. Lipid transfer between the inner and outer GUV leaflets occurs via a hemifusion pore that forms early in the process near the membrane contact site. This pore also permits expulsion of water from the GUV interior as the vesicle contracts onto the contact site. |
|
Coarse-Grained and Atomistic Simulations of the Salt-Stable Cowpea Chlorotic Mottle Virus (SS-CCMV) Subunit 26–49: β-Barrel Stability of the Hexamer and Pentamer Geometries
Tristan Bereau, Christoph Globisch, Markus Deserno, and Christine Peter
J. Chem. Theory Comput. 8, 3750–3758 (2012).
A combination of coarse-grained (CG) and atomistic simulations provides a suitable computational framework to study unstructured regions of proteins, for which experimental data are often lacking or limited. In this work, we combine CG and atomistic simulations with clustering algorithms and free energy reweighting methods to explore the conformational equilibrium of certain regions of the salt-stable cowpea chlorotic mottle virus (SS-CCMV). In particular, we focus on the geometry of converging strands (residues 26–49) from contacting subunits at the 3-fold (hexamer) and 5-fold (pentamer) symmetry points of the capsid. We show the following: (i) The simulations reproduce the experimentally observed β-barrel for the hexamer. (ii) The pentamer geometry is unable to stabilize a β-barrel conformation; it assumes various states instead, again in accordance with the experimental results which do not indicate a well-defined structure for the pentameric interface. (iii) Atomistic simulations of the backmapped CG structures remain relatively stable, indicative of plausible CG conformations and slow kinetics on the atomistic level. |
|
Determining the Gaussian curvature modulus in simulations
Mingyang Hu, John J. Briguglio, and Markus Deserno
Biophys. J. 102, 1403–1410 (2012).
The Gaussian curvature modulus κ' of lipid bilayers likely contributes more than 100 kcal/mol to every cellular fission or fusion event. This huge impact on membrane remodeling energetics might be a factor that co-determines the complex lipid composition of biomembranes through tuning of κ'. Yet, its value has been measured only for a handful of simple lipids, and no simulation has so far determined it better than a factor of two, rendering a systematic investigation of such enticing speculations impossible.
Here we propose a highly accurate method to determine κ' in computer simulations. It relies on the interplay between curvature stress and edge tension of partially curved axisymmetric membrane discs and requires determining their closing probability.
For a simplified lipid model we obtain κ' and its relation to the normal bending modulus κ for membranes differing both in stiffness and spontaneous lipid curvature. The elastic ratio κ'/κ can be determined with a few percent statistical accuracy. Its value agrees with the scarce experimental data, and its change with spontaneous lipid curvature is compatible with theoretical expectations, thereby granting additional information on monolayer properties. We also show that an alternative determination of these elastic parameters based on moments of the lateral stress profile gives markedly different and unphysical values. |
|
Effective field theory approach to fluctuation-induced forces between colloids at an interface
Cem Yolcu, Ira Z. Rothstein, and Markus Deserno
Phys. Rev. E 85, 011140 (2012).
We discuss an effective field theory (EFT) approach to the computation of fluctuation-induced interactions between particles bound to a thermally fluctuating fluid surface controlled by surface tension. By describing particles as points, EFT avoids computing functional integrals subject to difficult constraints. Still, all information pertaining to particle size and shape is systematically restored by amending the surface Hamiltonian with a derivative expansion. The free energy is obtained as a cumulant expansion, for which straightforward techniques exist. We derive a completedescription for rigid axisymmetric objects, which allows us to develop a full asymptotic expansion—in powers of the inverse distance—for the pair interaction. We also demonstrate by a few examples the efficiency with which multibody interactions can be computed. Moreover, although the main advantage of the EFT approach lies in explicit computation, we discuss how one can infer certain features of cases involving flexible or anisotropic objects. The EFT description also permits a systematic computation of ground-state surface-mediated interactions, which we illustrate with a few examples. |
|
2011 |
| |
Effective field theory approach to Casimir interactions on soft matter surfaces
Cem Yolcu, Ira Z. Rothstein, and Markus Deserno
Europhys. Lett. 96 20003 (2011).
A shorter version can also be found here: arXiv:1007.4760v2 [cond-mat.soft]
 We utilize an effective field theory approach to calculate Casimir interactions between
objects bound to thermally fluctuating fluid surfaces or interfaces. This approach circumvents
the complicated constraints imposed by such objects on the functional integration measure by
reverting to a point particle representation. To capture the finite-size effects, we augment the
Hamiltonian by a term ΔH that encapsulates the particles' response to external fields. ΔH is
systematically expanded in a series of terms, each of which scales homogeneously in the two power
counting parameters: λ = R/r, the ratio of the typical object size (R) to the typical distance
between them (r), and δ = kBT/k, where k is the modulus characterizing the surface energy. The
coefficients of the terms in ΔH correspond to generalized polarizabilities and thus the formalism
applies to rigid as well as deformable objects. We first illustrate and verify our approach by re-deriving known pair forces between circular objects bound to films or membranes. To demonstrate
its efficiency and versatility, we then derive a number of new results, among them the triplet
interactions present in these systems, a higher-order correction to the film interaction, and general
scaling laws for the leading-order interaction valid for objects of arbitrary shape and internal
flexibility.
We utilize an effective field theory approach to calculate Casimir interactions between
objects bound to thermally fluctuating fluid surfaces or interfaces. This approach circumvents
the complicated constraints imposed by such objects on the functional integration measure by
reverting to a point particle representation. To capture the finite-size effects, we augment the
Hamiltonian by a term ΔH that encapsulates the particles' response to external fields. ΔH is
systematically expanded in a series of terms, each of which scales homogeneously in the two power
counting parameters: λ = R/r, the ratio of the typical object size (R) to the typical distance
between them (r), and δ = kBT/k, where k is the modulus characterizing the surface energy. The
coefficients of the terms in ΔH correspond to generalized polarizabilities and thus the formalism
applies to rigid as well as deformable objects. We first illustrate and verify our approach by re-deriving known pair forces between circular objects bound to films or membranes. To demonstrate
its efficiency and versatility, we then derive a number of new results, among them the triplet
interactions present in these systems, a higher-order correction to the film interaction, and general
scaling laws for the leading-order interaction valid for objects of arbitrary shape and internal
flexibility.
|
|
Membrane mediated interactions between circular particles in the strongly curved regime
Benedict J. Reynwar and Markus Deserno
Soft Matter 7, 8567–8575 (2011).
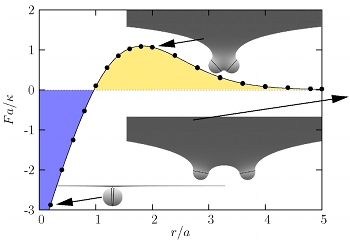 Particles binding to a fluid lipid membrane can induce bilayer deformations, for instance when these particles are curved.
Since the energy of two overlapping warp fields depends on the mutual distance between the two particles creating them, they will
experience forces mediated by the curvature of the membrane. If the deformations are sufficiently weak, the associated differential
equations for the membrane shape are linear, and the resulting interactions are understood very well; but for stronger curvature
imprint very little is known, owing to the highly nonlinear nature of the problem. Here we numerically calculate the magnitude
of such membrane-mediated interactions in the case of two axisymmetric particles over a wide range of curvature imprints, deep
into the nonlinear regime. We show that over an intermediate distance range the sign of the force reverses beyond a sufficiently
strong deformation. These findings are quantitatively confirmed by a simple analytical close-distance expansion. The sign flip
can be traced to a change in magnitude between the two principal curvatures midway between the two particles, which can only
occur at sufficient particle tilt, a condition which is by construction ruled out in the linearized description. We also show these
large perturbation results to agree with coarse-grained molecular dynamics simulations and suggest that a favorable comparison
is indeed more likely to hold in the strongly deformed regime.
Particles binding to a fluid lipid membrane can induce bilayer deformations, for instance when these particles are curved.
Since the energy of two overlapping warp fields depends on the mutual distance between the two particles creating them, they will
experience forces mediated by the curvature of the membrane. If the deformations are sufficiently weak, the associated differential
equations for the membrane shape are linear, and the resulting interactions are understood very well; but for stronger curvature
imprint very little is known, owing to the highly nonlinear nature of the problem. Here we numerically calculate the magnitude
of such membrane-mediated interactions in the case of two axisymmetric particles over a wide range of curvature imprints, deep
into the nonlinear regime. We show that over an intermediate distance range the sign of the force reverses beyond a sufficiently
strong deformation. These findings are quantitatively confirmed by a simple analytical close-distance expansion. The sign flip
can be traced to a change in magnitude between the two principal curvatures midway between the two particles, which can only
occur at sufficient particle tilt, a condition which is by construction ruled out in the linearized description. We also show these
large perturbation results to agree with coarse-grained molecular dynamics simulations and suggest that a favorable comparison
is indeed more likely to hold in the strongly deformed regime.
|
|
Structural basis of Folding Cooperativity in Model Proteins:
Insights from a Microcanonical Perspective
Tristan Bereau, Markus Deserno, and Michael Bachmann
Biophys. J. 100, 2764–2772 (2011).
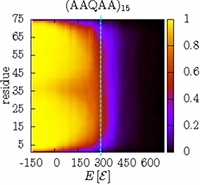 Two-state cooperativity is an important characteristic in protein folding. It is defined by a depletion of states that lie energetically between folded and unfolded conformations. There are different ways to test for two-state cooperativity; however, most of these approaches probe indirect proxies of this depletion. Generalized-ensemble computer simulations allow us to unambiguously identify this transition by a microcanonical analysis on the basis of the density of states. Here, we present a detailed characterization of several helical peptides obtained by coarse-grained simulations. The level of resolution of the coarse-grained model allowed to study realistic structures ranging from small α–helices to a de novo three-helix bundle without biasing the force field toward the native state of the protein. By linking thermodynamic and structural features, we are able to show that whereas short α–helices exhibit two-state cooperativity, the type of transition changes for longer chain lengths because the chain forms multiple helix nucleation sites, stabilizing a significant population of intermediate states. The helix bundle exhibits signs of two-state cooperativity owing to favorable helix-helix interactions, as predicted from theoretical models. A detailed analysis of secondary and tertiary structure formation fits well into the framework of several folding mechanisms and confirms features that up to now have been observed only in lattice models. Two-state cooperativity is an important characteristic in protein folding. It is defined by a depletion of states that lie energetically between folded and unfolded conformations. There are different ways to test for two-state cooperativity; however, most of these approaches probe indirect proxies of this depletion. Generalized-ensemble computer simulations allow us to unambiguously identify this transition by a microcanonical analysis on the basis of the density of states. Here, we present a detailed characterization of several helical peptides obtained by coarse-grained simulations. The level of resolution of the coarse-grained model allowed to study realistic structures ranging from small α–helices to a de novo three-helix bundle without biasing the force field toward the native state of the protein. By linking thermodynamic and structural features, we are able to show that whereas short α–helices exhibit two-state cooperativity, the type of transition changes for longer chain lengths because the chain forms multiple helix nucleation sites, stabilizing a significant population of intermediate states. The helix bundle exhibits signs of two-state cooperativity owing to favorable helix-helix interactions, as predicted from theoretical models. A detailed analysis of secondary and tertiary structure formation fits well into the framework of several folding mechanisms and confirms features that up to now have been observed only in lattice models.
|
|
2010 |
| |
Systematic implicit solvent coarse-graining of bilayer membranes:
lipid and phase transferability of the force field
Zun-Jing Wang and Markus Deserno
New Journal of Physics 12, 095004 (2010).
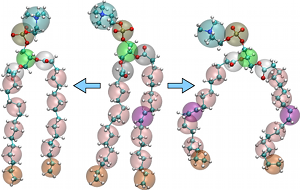 We study lipid and phase transferability of our recently developed systematically coarse-grained solvent-free membrane model. The force field was explicitly parametrized to describe a fluid 1-palmitoyl-2-oleoyl-phosphatidylcholine (POPC) bilayer at 310 K with correct structure and area per lipid, while gaining at least three orders of magnitude in computational effciency (see Ref. 1).
Here we show that exchanging CG tails, without any subsequent re-parameterization, creates reliable models of 1,2-dioleoylphosphatidylcholine (DOPC) and 1,2-dipalmitoylphosphatidylcholine
(DPPC) lipids in terms of structure and area per lipid. Furthermore, all CG lipids undergo a liquid-gel transition upon cooling, with characteristics as observed in experiments and all-atom
simulations during phase transformation. These studies suggest a promising transferability of our force field parameters to different lipid species and thermodynamic state-points, properties that
are prerequisite for even more complex systems, such as mixtures.
We study lipid and phase transferability of our recently developed systematically coarse-grained solvent-free membrane model. The force field was explicitly parametrized to describe a fluid 1-palmitoyl-2-oleoyl-phosphatidylcholine (POPC) bilayer at 310 K with correct structure and area per lipid, while gaining at least three orders of magnitude in computational effciency (see Ref. 1).
Here we show that exchanging CG tails, without any subsequent re-parameterization, creates reliable models of 1,2-dioleoylphosphatidylcholine (DOPC) and 1,2-dipalmitoylphosphatidylcholine
(DPPC) lipids in terms of structure and area per lipid. Furthermore, all CG lipids undergo a liquid-gel transition upon cooling, with characteristics as observed in experiments and all-atom
simulations during phase transformation. These studies suggest a promising transferability of our force field parameters to different lipid species and thermodynamic state-points, properties that
are prerequisite for even more complex systems, such as mixtures.
|
|
Interplay between secondary and tertiary structure formation in protein folding cooperativity
Tristan Bereau, Michael Bachmann, and Markus Deserno
J. Am. Chem. Soc. 132, 13129–13131 (2010).
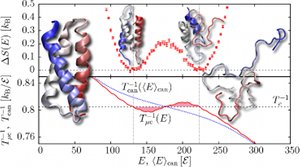 Protein folding cooperativity is defined by the nature of the finite-size thermodynamic transition exhibited upon folding: two-state transitions
show a free energy barrier between the folded and unfolded ensembles, while downhill folding is barrierless. A microcanonical analysis,
where the energy is the natural variable, has shown better suited to unambiguously characterize the nature of the transition compared to its
canonical counterpart. Replica exchange molecular dynamics simulations of a high resolution coarse-grained model allow for the accurate
evaluation of the density of states, in order to extract precise thermodynamic information, and measure its impact on structural features. The
method is applied to three helical peptides: a short helix shows sharp features of a two-state folder, while a longer helix and a three-helix
bundle exhibit downhill and two-state transitions, respectively. Extending the results of lattice simulations and theoretical models, we find
that it is the interplay between secondary structure and the loss of non-native tertiary contacts which determines the nature of the transition.
Protein folding cooperativity is defined by the nature of the finite-size thermodynamic transition exhibited upon folding: two-state transitions
show a free energy barrier between the folded and unfolded ensembles, while downhill folding is barrierless. A microcanonical analysis,
where the energy is the natural variable, has shown better suited to unambiguously characterize the nature of the transition compared to its
canonical counterpart. Replica exchange molecular dynamics simulations of a high resolution coarse-grained model allow for the accurate
evaluation of the density of states, in order to extract precise thermodynamic information, and measure its impact on structural features. The
method is applied to three helical peptides: a short helix shows sharp features of a two-state folder, while a longer helix and a three-helix
bundle exhibit downhill and two-state transitions, respectively. Extending the results of lattice simulations and theoretical models, we find
that it is the interplay between secondary structure and the loss of non-native tertiary contacts which determines the nature of the transition.
|
|
Cell Model Approach to Membrane Mediated Protein Interactions
Martin M. Müller and Markus Deserno
Progr. Theor. Phys. Suppl. 184, 351–363 (2010).
 Membrane-deforming proteins can interact through the curvature fields they create. In the case of many such proteins a cell model approach can be used to calculate the energy per protein and predict, whether it would lead to phase segregation or bud-formation. Using covariant differential geometry exact results are derived for the lateral pressure in terms of geometric properties at the cell boundary. Numerical solutions of the exact shape equations in the highly nonlinear regime are found and it is seen that both phase segregation and bud formation can occur.
Membrane-deforming proteins can interact through the curvature fields they create. In the case of many such proteins a cell model approach can be used to calculate the energy per protein and predict, whether it would lead to phase segregation or bud-formation. Using covariant differential geometry exact results are derived for the lateral pressure in terms of geometric properties at the cell boundary. Numerical solutions of the exact shape equations in the highly nonlinear regime are found and it is seen that both phase segregation and bud formation can occur.
|
|
A systematically coarse-grained solvent-free model for quantitative phospholipid bilayer simulations
Zun-Jing Wang and Markus Deserno
J. Phys. Chem. B 114, 11207 (2010).
 We presented an implicit solvent coarse-grained (CG) model for quantitative simulations of We presented an implicit solvent coarse-grained (CG) model for quantitative simulations of
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) bilayers. The use of implicit solvent enables membrane simulations on large length- and time-scales at moderate computational expense. Despite an improved computational efficiency, the model preserves chemical specificity and quantitative accuracy. The bonded and nonbonded interactions together with the effective cohesion mimicking the hydrophobic effect were systematically tuned by matching structural and mechanical properties from experiments and all-atom bilayer simulations, such as saturated area per lipid, radial distribution functions, density and pressure profiles across the bilayer, P2 order, etc. The CG lipid model is shown to self-assemble into a bilayer starting from a random dispersion. Its elastic properties, such as bending and stretching modulus, are semi-quantitatively consistent with experiments. The effects of (i) reduced molecular friction and (ii) more efficient integration combine to an overall speedup of three to four orders of magnitude compared to all-atom bilayer simulations. Our CG lipid model is especially useful for studies of large-scale phenomena in membranes which nevertheless require a fair description of chemical specificity, e.g. membrane patches interacting with movable and transformable membrane proteins and peptides.
|
|
In-plane homogeneity and lipid dynamics in tethered Bilayer Lipid Membranes (tBLMs)
Siddharth Shenoy, Radu Moldovan, James Fitzpatrick, David J. Vanderah, Markus Deserno, and Mathias Lösche
Soft Matter 6, 1263 (2010).
 Tethered bilayer lipid membranes (tBLMs) were prepared by the self-assembly of thiolated lipidic anchor molecules on gold, followed by phospholipid precipitation via rapid solvent exchange. They were characterized in their in-plane structure, dynamics and dielectric properties. We find that the in-plane homogeneity and resistivity of the tBLMs depends critically on a well-controlled sample environment during the rapid solvent exchange procedure. The in-plane dynamics of the systems, assessed by fluorescence correlation spectroscopy (FCS) as the diffusivity of free, labeled phospholipid dissolved in the membrane, depends on the density of the lipidic anchors in the bilayer leaflet proximal to the substrate as well as on details of the molecular structure of the anchor lipid. In DOPC tBLMs in which tethers are laterally dilute (sparsely-tethered bilayer lipid membranes, stBLMs), measured diffusivities, D ~ 4 μm2/s, are only slightly greater than those reported in physisorbed bilayers. However, when we distinguish label diffusion in the proximal and in the distal bilayer leaflets, we observe distinct diffusivities, D ~ 2 μm2/s and ~ 7 μm2/s, respectively. The value observed in the distal leaflet is identical to that in free membranes. stBLMs completed with phytanoyl lipids (DPhyPC) show consistently lower label diffusivity than those completed with unsaturated chains (DOPC). As the flexibility of the tether chain increases, a reduction in the apparent diffusivityis observed, which we interpret as an increased propensity of the proximal bilayer leaflet to host free lipid. Finally, in laterally heterogeneous bilayers, the label diffusivity varies only by a factor of ~ 2× – 4×, indicating that the distinct regions in the bilayers do not correspond to distinct phases such as a fluid phase coexisting with a gel phase. Tethered bilayer lipid membranes (tBLMs) were prepared by the self-assembly of thiolated lipidic anchor molecules on gold, followed by phospholipid precipitation via rapid solvent exchange. They were characterized in their in-plane structure, dynamics and dielectric properties. We find that the in-plane homogeneity and resistivity of the tBLMs depends critically on a well-controlled sample environment during the rapid solvent exchange procedure. The in-plane dynamics of the systems, assessed by fluorescence correlation spectroscopy (FCS) as the diffusivity of free, labeled phospholipid dissolved in the membrane, depends on the density of the lipidic anchors in the bilayer leaflet proximal to the substrate as well as on details of the molecular structure of the anchor lipid. In DOPC tBLMs in which tethers are laterally dilute (sparsely-tethered bilayer lipid membranes, stBLMs), measured diffusivities, D ~ 4 μm2/s, are only slightly greater than those reported in physisorbed bilayers. However, when we distinguish label diffusion in the proximal and in the distal bilayer leaflets, we observe distinct diffusivities, D ~ 2 μm2/s and ~ 7 μm2/s, respectively. The value observed in the distal leaflet is identical to that in free membranes. stBLMs completed with phytanoyl lipids (DPhyPC) show consistently lower label diffusivity than those completed with unsaturated chains (DOPC). As the flexibility of the tether chain increases, a reduction in the apparent diffusivityis observed, which we interpret as an increased propensity of the proximal bilayer leaflet to host free lipid. Finally, in laterally heterogeneous bilayers, the label diffusivity varies only by a factor of ~ 2× – 4×, indicating that the distinct regions in the bilayers do not correspond to distinct phases such as a fluid phase coexisting with a gel phase.
|
|
2009 |
| |
Generic coarse-grained model for protein folding and aggregation
Tristan Bereau and Markus Deserno
J. Chem. Phys. 130, 235106 (2009).
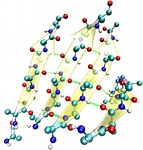 A generic coarse-grained (CG) protein model is presented. The intermediate level of resolution (four beads per amino acid, implicit solvent) allows for accurate sampling of local conformations. It relies on simple interactions that emphasize structure, such as hydrogen bonds and hydrophobicity. Realistic alpha/beta content is achieved by including an effective nearest-neighbor dipolar interaction. Parameters are tuned to reproduce both local conformations and tertiary structures. The thermodynamics and kinetics of a three-helix bundle are studied. We check that the CG model is able to fold proteins with tertiary structures and amino acid sequences different from the one used for parameter tuning. By studying both helical and extended conformations we make sure the force field is not biased toward any particular secondary structure. The accuracy involved in folding not only the test protein but also other ones show strong evidence for amino acid cooperativity embedded in the model. Without any further adjustments or bias a realistic oligopeptide aggregation scenario is observed. A generic coarse-grained (CG) protein model is presented. The intermediate level of resolution (four beads per amino acid, implicit solvent) allows for accurate sampling of local conformations. It relies on simple interactions that emphasize structure, such as hydrogen bonds and hydrophobicity. Realistic alpha/beta content is achieved by including an effective nearest-neighbor dipolar interaction. Parameters are tuned to reproduce both local conformations and tertiary structures. The thermodynamics and kinetics of a three-helix bundle are studied. We check that the CG model is able to fold proteins with tertiary structures and amino acid sequences different from the one used for parameter tuning. By studying both helical and extended conformations we make sure the force field is not biased toward any particular secondary structure. The accuracy involved in folding not only the test protein but also other ones show strong evidence for amino acid cooperativity embedded in the model. Without any further adjustments or bias a realistic oligopeptide aggregation scenario is observed.
|
|
Multiscale modeling of emergent materials: biological and soft matter
Teemu Murtola, Alex Bunker, Ilpo Vattulainen, Markus Deserno, Mikko Karttunen
Phys. Chem. Chem. Phys. 11, 1869–1892 (2009).
In this review, we focus on four current related issues in multiscale modeling of soft and biological matter. First, we discuss how to use structural information from detailed models (or experiments) to construct coarse-grained ones in a hierarchical and systematic way. This is discussed in the context of the so-called Henderson theorem and the inverse Monte Carlo method of Lyubartsev and Laaksonen. In the second part, we take a different look at coarse graining by analyzing conformations of molecules. This is done by the application of self-organizing maps, i.e., a neural network type approach. Such an approach can be used to guide the selection of the relevant degrees of freedom. Then, we discuss technical issues related to the popular dissipative particle dynamics (DPD) method. Importantly, the potentials derived using the inverse Monte Carlo method can be used together with the DPD thermostat. In the final part we focus on solvent-free modeling which offers a different route to coarse graining by integrating out the degrees of freedom associated with solvent. |
|
Mesoscopic Membrane Physics: Concepts, Simulations, and Selected Applications
Markus Deserno
Macromol. Rapid. Comm. 30, 752–771 (2009).
 The window of a few tens to a few hundred nanometers in length scale is a booming field in lipid membrane research, owing largely to two reasons. First, many exciting biophysical and cell biological processes take place within it. Second, experimental techniques manage to zoom in on this sub-optical scale, while computer simulations zoom out to system sizes previously unattainable, and both will be meeting soon. This paper reviews a selection of questions and concepts in this field and demonstrates that they can often be favorably addressed with highly simplified simulation models. Among the topics discussed are membrane adhesion to substrates, mixed lipid bilayers, lipid curvature coupling, pore formation by antimicrobial peptides, composition-driven protein aggregation, and curvature driven vesiculation. The window of a few tens to a few hundred nanometers in length scale is a booming field in lipid membrane research, owing largely to two reasons. First, many exciting biophysical and cell biological processes take place within it. Second, experimental techniques manage to zoom in on this sub-optical scale, while computer simulations zoom out to system sizes previously unattainable, and both will be meeting soon. This paper reviews a selection of questions and concepts in this field and demonstrates that they can often be favorably addressed with highly simplified simulation models. Among the topics discussed are membrane adhesion to substrates, mixed lipid bilayers, lipid curvature coupling, pore formation by antimicrobial peptides, composition-driven protein aggregation, and curvature driven vesiculation.
|
|
2008 |
| |
Membrane composition-mediated protein-protein interactions
Benedict J. Reynwar, Markus Deserno
Biointerphases 3, FA117 (2008).
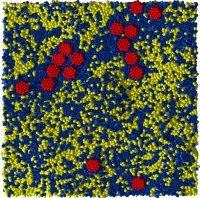 The authors investigate membrane composition-mediated interactions between proteins adsorbed onto a two-component lipid bilayer close to critical demixing using coarse-grained molecular dynamics simulations and a phenomenological Ginzburg-Landau theory. The simulations consist of three-bead lipids and platelike proteins, which adsorb onto the membrane by binding preferentially to one of the two lipid species. The composition profile around one protein and the pair correlation function between two proteins are measured and compared to the analytical predictions. The theoretical framework is applicable to any scalar field embedded in the membrane, and although in this work the authors treat flat membranes, the methodology extends readily to curved geometries. Neglecting fluctuations, both lipid composition profile and induced protein pair potential are predicted to follow a zeroth order modified Bessel function of the second kind with the same characteristic decay length. These predictions are consistent with our molecular dynamics simulations, except that the interaction range is found to be larger than the single profile correlation length. The authors investigate membrane composition-mediated interactions between proteins adsorbed onto a two-component lipid bilayer close to critical demixing using coarse-grained molecular dynamics simulations and a phenomenological Ginzburg-Landau theory. The simulations consist of three-bead lipids and platelike proteins, which adsorb onto the membrane by binding preferentially to one of the two lipid species. The composition profile around one protein and the pair correlation function between two proteins are measured and compared to the analytical predictions. The theoretical framework is applicable to any scalar field embedded in the membrane, and although in this work the authors treat flat membranes, the methodology extends readily to curved geometries. Neglecting fluctuations, both lipid composition profile and induced protein pair potential are predicted to follow a zeroth order modified Bessel function of the second kind with the same characteristic decay length. These predictions are consistent with our molecular dynamics simulations, except that the interaction range is found to be larger than the single profile correlation length.
|
|
Coarse-grained modeling of interactions of lipid bilayers with supports
Matthew I. Hoopes, Markus Deserno, Margie L. Longo, Roland Faller
J. Chem. Phys. 129, 175102 (2008).
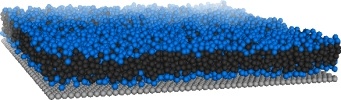 We characterize the differences between supported and unsupported lipid bilayer membranes using a mesoscopic simulation model and a simple particle-based realization for a flat support on to which the lipids are adsorbed. We show that the nanometer roughness of the support affects membrane binding strength very little. We then compare the lipid distributions and pressure profiles of free and supported membranes. The surface localization of the proximal leaflet breaks the symmetry seen in a free bilayer, and we quantify the entropic penalty for binding and the increased lateral compression modulus. We characterize the differences between supported and unsupported lipid bilayer membranes using a mesoscopic simulation model and a simple particle-based realization for a flat support on to which the lipids are adsorbed. We show that the nanometer roughness of the support affects membrane binding strength very little. We then compare the lipid distributions and pressure profiles of free and supported membranes. The surface localization of the proximal leaflet breaks the symmetry seen in a free bilayer, and we quantify the entropic penalty for binding and the increased lateral compression modulus.
|
|
Adhesion promotes phase separation in mixed-lipid membranes
Vernita D. Gordon, Markus Deserno, Carolyn M. J. Andrew, Stefan U. Egelhaaf, Wilson C. K. Poon
Europhys. Lett. 84, 48003 (2008).
 We investigate the interplay of domain formation and adhesion in mixed-lipid membranes. Giant unilamellar vesicles consisting of two- and three-component lipid mixtures are studied using confocal fluorescence microscopy. Upon driving the system towards the demixing transition, phase separation is invariably found to occur first in regions where membranes adhere to one another, despite identical lipid headgroups and negligible curvature effects. We propose a simple generic mechanism based on the suppression of thermal shape fluctuations to explain these observations. Our findings suggest novel possibilities by which biomembranes can create and utilize lateral lipid heterogeneities. We investigate the interplay of domain formation and adhesion in mixed-lipid membranes. Giant unilamellar vesicles consisting of two- and three-component lipid mixtures are studied using confocal fluorescence microscopy. Upon driving the system towards the demixing transition, phase separation is invariably found to occur first in regions where membranes adhere to one another, despite identical lipid headgroups and negligible curvature effects. We propose a simple generic mechanism based on the suppression of thermal shape fluctuations to explain these observations. Our findings suggest novel possibilities by which biomembranes can create and utilize lateral lipid heterogeneities.
|
|
Coarse-Grained Simulation Studies of Peptide-Induced Pore Formation
Gregoria Illya, Markus Deserno
Biophys. J. 95, 4163–4173 (2008).
 We investigate the interactions between lipid bilayers and amphiphilic peptides using a solvent-free coarse-grained simulation technique. In our model, each lipid is represented by one hydrophilic and three hydrophobic beads. The amphiphilic peptide is modeled as a hydrophobic-hydrophilic cylinder with hydrophilic caps. We find that with increasing peptide-lipid attraction the preferred state of the peptide changes from desorbed, to adsorbed, to inserted. A single peptide with weak attraction binds on the bilayer surface, while one with strong attraction spontaneously inserts into the bilayer. We show how several peptides, which individually bind only to the bilayer surface, cooperatively insert. Furthermore, hydrophilic strips along the peptide cylinder induce the formation of multipeptide pores, whose size and morphology depend on the peptides' overall hydrophilicity, the distribution of hydrophilic residues, and the peptide-peptide interactions. Strongly hydrophilic peptides insert less readily, but prove to be more destructive to bilayer integrity. We investigate the interactions between lipid bilayers and amphiphilic peptides using a solvent-free coarse-grained simulation technique. In our model, each lipid is represented by one hydrophilic and three hydrophobic beads. The amphiphilic peptide is modeled as a hydrophobic-hydrophilic cylinder with hydrophilic caps. We find that with increasing peptide-lipid attraction the preferred state of the peptide changes from desorbed, to adsorbed, to inserted. A single peptide with weak attraction binds on the bilayer surface, while one with strong attraction spontaneously inserts into the bilayer. We show how several peptides, which individually bind only to the bilayer surface, cooperatively insert. Furthermore, hydrophilic strips along the peptide cylinder induce the formation of multipeptide pores, whose size and morphology depend on the peptides' overall hydrophilicity, the distribution of hydrophilic residues, and the peptide-peptide interactions. Strongly hydrophilic peptides insert less readily, but prove to be more destructive to bilayer integrity.
|
|
2007 |
| |
Aggregation and vesiculation of membrane proteins by curvature-mediated interactions
Benedict J. Reynwar, Gregoria Illya, Vagelis A. Harmandaris, Martin M. Müller, Kurt Kremer, and Markus Deserno
Nature 447, 461–464 (2007).
 Membrane remodelling plays an important role in cellular tasks such as endocytosis, vesiculation and protein sorting, and in the biogenesis of organelles such as the endoplasmic reticulum or the Golgi apparatus. It is well established that the remodelling process is aided by specialized proteins that can sense as well as create membrane curvature, and trigger tubulation when added to synthetic liposomes. Because the energy needed for such large-scale changes in membrane geometry significantly exceeds the binding energy between individual proteins and between protein and membrane, cooperative action is essential. It has recently been suggested that curvature-mediated attractive interactions could aid cooperation and complement the effects of specific binding events on membrane remodelling. But it is difficult to experimentally isolate curvature-mediated interactions from direct attractions between proteins. Moreover, approximate theories predict repulsion between isotropically curving proteins. Here we use coarse-grained membrane simulations to show that curvature-inducing model proteins adsorbed on lipid bilayer membranes can experience attractive interactions that arise purely as a result of membrane curvature. We find that once a minimal local bending is realized, the effect robustly drives protein cluster formation and subsequent transformation into vesicles with radii that correlate with the local curvature imprint. Owing to its universal nature, curvature-mediated attraction can operate even between proteins lacking any specific interactions, such as newly synthesized and still immature membrane proteins in the endoplasmic reticulum. Membrane remodelling plays an important role in cellular tasks such as endocytosis, vesiculation and protein sorting, and in the biogenesis of organelles such as the endoplasmic reticulum or the Golgi apparatus. It is well established that the remodelling process is aided by specialized proteins that can sense as well as create membrane curvature, and trigger tubulation when added to synthetic liposomes. Because the energy needed for such large-scale changes in membrane geometry significantly exceeds the binding energy between individual proteins and between protein and membrane, cooperative action is essential. It has recently been suggested that curvature-mediated attractive interactions could aid cooperation and complement the effects of specific binding events on membrane remodelling. But it is difficult to experimentally isolate curvature-mediated interactions from direct attractions between proteins. Moreover, approximate theories predict repulsion between isotropically curving proteins. Here we use coarse-grained membrane simulations to show that curvature-inducing model proteins adsorbed on lipid bilayer membranes can experience attractive interactions that arise purely as a result of membrane curvature. We find that once a minimal local bending is realized, the effect robustly drives protein cluster formation and subsequent transformation into vesicles with radii that correlate with the local curvature imprint. Owing to its universal nature, curvature-mediated attraction can operate even between proteins lacking any specific interactions, such as newly synthesized and still immature membrane proteins in the endoplasmic reticulum.
|
|
Contact lines for fluid surface adhesion
Martin M. Müller, M. Deserno, and J. Guven
Phys. Rev. E 76, 011605 (2007); see also cond-mat/0703019
 When a fluid surface adheres to a substrate, the location of the contact line adjusts in order to minimize the overall energy. This implies boundary conditions which depend on the characteristic surface deformation energies. We develop a general geometrical framework within which these conditions can be derived in a completely systematic way. We treat both adhesion to a rigid substrate and adhesion between two fluid surfaces, and illustrate our general results for several important Hamiltonians involving both curvature and curvature gradients. Some of these have previously been studied using very different techniques. With the exception of capillary phenomena, the Hamiltonian will not only be sensitive to boundary translations, but may also respond to changes in slope and even in curvature. The functional form of the additional contributions will follow readily from our treatment. We will show that the boundary conditions describing adhesion between two fluid surfaces express the balance of stresses and torques, as one would expect. At a rigid substrate, however, this simple identification will generally fail. This is because local rotations of the surface normal will be entirely "enslaved" to translations on the substrate. As a consequence, stresses and torques enter a single balance condition and cannot be disentangled. When a fluid surface adheres to a substrate, the location of the contact line adjusts in order to minimize the overall energy. This implies boundary conditions which depend on the characteristic surface deformation energies. We develop a general geometrical framework within which these conditions can be derived in a completely systematic way. We treat both adhesion to a rigid substrate and adhesion between two fluid surfaces, and illustrate our general results for several important Hamiltonians involving both curvature and curvature gradients. Some of these have previously been studied using very different techniques. With the exception of capillary phenomena, the Hamiltonian will not only be sensitive to boundary translations, but may also respond to changes in slope and even in curvature. The functional form of the additional contributions will follow readily from our treatment. We will show that the boundary conditions describing adhesion between two fluid surfaces express the balance of stresses and torques, as one would expect. At a rigid substrate, however, this simple identification will generally fail. This is because local rotations of the surface normal will be entirely "enslaved" to translations on the substrate. As a consequence, stresses and torques enter a single balance condition and cannot be disentangled.
|
|
Balancing torques in membrane-mediated interactions: exact results and numerical illustrations
Martin M. Müller, M. Deserno, and J. Guven
Phys. Rev. E 76, 011921 (2007); see also cond-mat/0702340.
 Torques on interfaces can be described by a divergence-free tensor which is fully encoded in the geometry. This tensor consists of two terms, one originating in the couple of the stress, the other capturing an intrinsic contribution due to curvature. In analogy to the description of forces in terms of a stress tensor, the torque on a particle can be expressed as a line integral along a contour surrounding the particle. Interactions between particles mediated by a fluid membrane are studied within this framework. In particular, torque balance places a strong constraint on the shape of the membrane. Symmetric two-particle configurations admit simple analytical expressions which are valid in the fully nonlinear regime; in particular, the problem may be solved exactly in the case of two membrane-bound parallel cylinders. This apparently simple system provides some flavor of the remarkably subtle nonlinear behavior associated with membrane-mediated interactions. Torques on interfaces can be described by a divergence-free tensor which is fully encoded in the geometry. This tensor consists of two terms, one originating in the couple of the stress, the other capturing an intrinsic contribution due to curvature. In analogy to the description of forces in terms of a stress tensor, the torque on a particle can be expressed as a line integral along a contour surrounding the particle. Interactions between particles mediated by a fluid membrane are studied within this framework. In particular, torque balance places a strong constraint on the shape of the membrane. Symmetric two-particle configurations admit simple analytical expressions which are valid in the fully nonlinear regime; in particular, the problem may be solved exactly in the case of two membrane-bound parallel cylinders. This apparently simple system provides some flavor of the remarkably subtle nonlinear behavior associated with membrane-mediated interactions.
|
|
2006 |
| |
A novel method for measuring the bending rigidity of model lipid membranes by simulating tethers
Vagelis A. Harmandaris and Markus Deserno
J. Chem. Phys. 125, 204905 (2006); see also cond-mat/0611012.
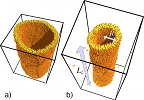 The tensile force along a cylindrical lipid bilayer tube is proportional to the membrane's bending modulus and inversely proportional to the tube radius. We show that this relation, which is experimentally exploited to measure bending rigidities, can be applied with even greater ease in computer simulations. Using a coarse-grained bilayer model we efficiently obtain bending rigidities that compare very well with complementary measurements based on an analysis of thermal undulation modes. We furthermore illustrate that no deviations from simple quadratic continuum theory occur up to a radius of curvature comparable to the bilayer thickness. The tensile force along a cylindrical lipid bilayer tube is proportional to the membrane's bending modulus and inversely proportional to the tube radius. We show that this relation, which is experimentally exploited to measure bending rigidities, can be applied with even greater ease in computer simulations. Using a coarse-grained bilayer model we efficiently obtain bending rigidities that compare very well with complementary measurements based on an analysis of thermal undulation modes. We furthermore illustrate that no deviations from simple quadratic continuum theory occur up to a radius of curvature comparable to the bilayer thickness.
|
|
How to determine local elastic properties of lipid bilayer membranes from atomic-force-microscope measurements: A theoretical analysis
Davood Norouzi, Martin M. Müller, and Markus Deserno
Phys. Rev. E 74, 061914 (2006); see also cond-mat/0602662.
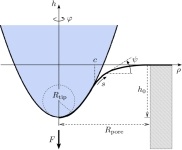 Measurements with an atomic force microscope (AFM) offer a direct way to probe elastic properties of lipid bilayer membranes locally: provided the underlying stress-strain relation is known, material parameters such as surface tension or bending rigidity may be deduced. In a recent experiment a pore-spanning membrane was poked with an AFM tip, yielding a linear behavior of the force-indentation curves. A theoretical model for this case is presented here which describes these curves in the framework of Helfrich theory. The linear behavior of the measurements is reproduced if one neglects the influence of adhesion between tip and membrane. Including it via an adhesion balance changes the situation significantly: force-distance curves cease to be linear, hysteresis and nonzero detachment forces can show up. The characteristics of this rich scenario are discussed in detail in this paper. Measurements with an atomic force microscope (AFM) offer a direct way to probe elastic properties of lipid bilayer membranes locally: provided the underlying stress-strain relation is known, material parameters such as surface tension or bending rigidity may be deduced. In a recent experiment a pore-spanning membrane was poked with an AFM tip, yielding a linear behavior of the force-indentation curves. A theoretical model for this case is presented here which describes these curves in the framework of Helfrich theory. The linear behavior of the measurements is reproduced if one neglects the influence of adhesion between tip and membrane. Including it via an adhesion balance changes the situation significantly: force-distance curves cease to be linear, hysteresis and nonzero detachment forces can show up. The characteristics of this rich scenario are discussed in detail in this paper.
|
|
Mechanical properties of pore-spanning lipid bilayers probed by atomic force microscopy
Siegfried Steltenkamp, Martin M. Müller, Markus Deserno, Christian Hennesthal, Claudia Steinem, and Andreas Janshoff
Biophys. J. 91, 217–226 (2006).
 We measure the elastic response of a free-standing lipid membrane to a local indentation by using an atomic force microscope (AFM). Starting point is a planar goldcoated alumina substrate with a chemisorbed 3-mercaptopropionic acid monolayer displaying circular pores of very well defined and tunable size, over which N,N,-dimethyl-N,N,- dioctadecylammonium bromide or 1,2-dioleoyl-3-trimethylammonium-propane chloride were spread. Centrally indenting these "nanodrums" with an AFM tip yields force-indentation curves, which we quantitatively analyze by solving the corresponding shape equations of continuum curvature elasticity. Since the measured response depends in a known way on the system geometry (pore size, tip radius) and on material parameters (bending modulus, lateral tension), this opens the possibility to monitor local elastic properties of lipid membranes in a well-controlled setting. We measure the elastic response of a free-standing lipid membrane to a local indentation by using an atomic force microscope (AFM). Starting point is a planar goldcoated alumina substrate with a chemisorbed 3-mercaptopropionic acid monolayer displaying circular pores of very well defined and tunable size, over which N,N,-dimethyl-N,N,- dioctadecylammonium bromide or 1,2-dioleoyl-3-trimethylammonium-propane chloride were spread. Centrally indenting these "nanodrums" with an AFM tip yields force-indentation curves, which we quantitatively analyze by solving the corresponding shape equations of continuum curvature elasticity. Since the measured response depends in a known way on the system geometry (pore size, tip radius) and on material parameters (bending modulus, lateral tension), this opens the possibility to monitor local elastic properties of lipid membranes in a well-controlled setting.
|
|
Coupling between lipid shape and membrane curvature
Ira R. Cooke and Markus Deserno
Biophys. J. 91, 487–495 (2006).
 Using molecular dynamics simulations, we examine the behavior of lipids whose preferred curvature can be systematically varied. This curvature is imposed by controlling the head group size of a coarse grained lipid model recently developed by us. In order to validate this approach we examine self assembly of each individual lipid type and observe the complete range of expected bilayer and micelle phases. We then examine binary systems consisting of lipids with positive and negative preferred curvature and find a definite sorting effect. Lipids with positive preferred curvature are found in greater proportions in outer monolayers with the opposite observed for lipids with negative preferred curvature. We also observe a similar, but slightly stronger effect for lipids in a developing spherical bud formed by adhesion to a colloid (eg a viral capsid). Importantly, the magnitude of this effect in both cases was large only for regions with strong mean curvature (radii of curvature <10nm). Our results suggest that lipid shape must act in concert with other physico-chemical effects such as phase transitions or interactions with proteins to produce strong sorting in cellular pathways. Using molecular dynamics simulations, we examine the behavior of lipids whose preferred curvature can be systematically varied. This curvature is imposed by controlling the head group size of a coarse grained lipid model recently developed by us. In order to validate this approach we examine self assembly of each individual lipid type and observe the complete range of expected bilayer and micelle phases. We then examine binary systems consisting of lipids with positive and negative preferred curvature and find a definite sorting effect. Lipids with positive preferred curvature are found in greater proportions in outer monolayers with the opposite observed for lipids with negative preferred curvature. We also observe a similar, but slightly stronger effect for lipids in a developing spherical bud formed by adhesion to a colloid (eg a viral capsid). Importantly, the magnitude of this effect in both cases was large only for regions with strong mean curvature (radii of curvature <10nm). Our results suggest that lipid shape must act in concert with other physico-chemical effects such as phase transitions or interactions with proteins to produce strong sorting in cellular pathways.
|
|
2005 |
| |
Solvent free model for self-assembling fluid bilayer membranes: Stabilization of the fluid phase based on broad attractive tail potentials
Ira R. Cooke and Markus Deserno
J. Chem. Phys. 123, 224710 (2005).
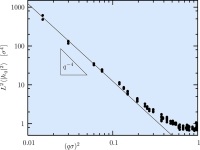 We present a simple and highly adaptable method for simulating coarse-grained lipid membranes without explicit solvent. Lipids are represented by one head-bead and two tail-beads, with the interaction between tails being of key importance in stabilizing the fluid phase. Two such tail-tail potentials were tested, with the important feature in both cases being a variable range of attraction. We examined phase diagrams of this range versus temperature for both functional forms of the tail-tail attraction and found that a certain threshold attractive width was required to stabilize the fluid phase. Within the fluid phase region we find that material properties such as area per lipid, orientational order, diffusion constant, inter-leaflet flip-flop rate and bilayer stiffness all depend strongly and monotonically on the attractive width. For three particular values of the potential width we investigate the transition between gel and fluid phases via heating or cooling and find that this transition is discontinuous with considerable hysteresis. We also investigated the stretching of a bilayer to eventually form a pore and found excellent agreement with recent analytic theory. We present a simple and highly adaptable method for simulating coarse-grained lipid membranes without explicit solvent. Lipids are represented by one head-bead and two tail-beads, with the interaction between tails being of key importance in stabilizing the fluid phase. Two such tail-tail potentials were tested, with the important feature in both cases being a variable range of attraction. We examined phase diagrams of this range versus temperature for both functional forms of the tail-tail attraction and found that a certain threshold attractive width was required to stabilize the fluid phase. Within the fluid phase region we find that material properties such as area per lipid, orientational order, diffusion constant, inter-leaflet flip-flop rate and bilayer stiffness all depend strongly and monotonically on the attractive width. For three particular values of the potential width we investigate the transition between gel and fluid phases via heating or cooling and find that this transition is discontinuous with considerable hysteresis. We also investigated the stretching of a bilayer to eventually form a pore and found excellent agreement with recent analytic theory.
|
|
Interface mediated interactions between particles—a geometrical approach
Martin M. Müller, M. Deserno, and J. Guven
Phys. Rev. E 72, 061407 (2005).
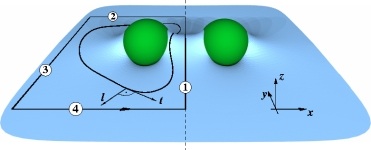 Particles bound to an interface interact because they deform its shape. The stresses that result are fully encoded in the geometry and described by a divergence-free surface stress tensor. This stress tensor can be used to express the force on a particle as a line integral along any conveniently chosen closed contour that surrounds the particle. The resulting expression is exact (i.e., free of any "smallness" assumptions) and independent of the chosen surface parametrization. Additional surface degrees of freedom, such as vector fields describing lipid tilt, are readily included in this formalism. As an illustration, we derive the exact force for several important surface Hamiltonians in various symmetric two-particle configurations in terms of the midplane geometry; its sign is evident in certain interesting limits. Specializing to the linear regime, where the shape can be analytically determined, these general expressions yield force-distance relations, several of which have originally been derived by using an energy based approach. Particles bound to an interface interact because they deform its shape. The stresses that result are fully encoded in the geometry and described by a divergence-free surface stress tensor. This stress tensor can be used to express the force on a particle as a line integral along any conveniently chosen closed contour that surrounds the particle. The resulting expression is exact (i.e., free of any "smallness" assumptions) and independent of the chosen surface parametrization. Additional surface degrees of freedom, such as vector fields describing lipid tilt, are readily included in this formalism. As an illustration, we derive the exact force for several important surface Hamiltonians in various symmetric two-particle configurations in terms of the midplane geometry; its sign is evident in certain interesting limits. Specializing to the linear regime, where the shape can be analytically determined, these general expressions yield force-distance relations, several of which have originally been derived by using an energy based approach.
|
|
Tunable generic model for fluid bilayer membranes
Ira R. Cooke, Kurt Kremer, and Markus Deserno
Phys. Rev. E 72, 011506 (2005).
 We present a model for the efficient simulation of generic bilayer membranes. Individual lipids are represented by one head bead and two tail-beads. By means of simple pair potentials these robustly self-assemble to a fluid bilayer state over a wide range of parameters, without the need for an explicit solvent. The model shows the expected elastic behavior on large length scales, and its physical properties (e.g. fluidity or bending stiffness) can be widely tuned via a single parameter. In particular, bending rigidities in the experimentally relevant range are obtained, at least within 3–30kT. The model is naturally suited to study many physical topics, including self-assembly, fusion, bilayer melting, lipid mixtures, rafts, and protein-bilayer interactions. We present a model for the efficient simulation of generic bilayer membranes. Individual lipids are represented by one head bead and two tail-beads. By means of simple pair potentials these robustly self-assemble to a fluid bilayer state over a wide range of parameters, without the need for an explicit solvent. The model shows the expected elastic behavior on large length scales, and its physical properties (e.g. fluidity or bending stiffness) can be widely tuned via a single parameter. In particular, bending rigidities in the experimentally relevant range are obtained, at least within 3–30kT. The model is naturally suited to study many physical topics, including self-assembly, fusion, bilayer melting, lipid mixtures, rafts, and protein-bilayer interactions.
|
|
Geometry of surface mediated interactions
Martin M. Müller, M. Deserno, and J. Guven
Europhys. Lett. 69, 482–488 (2005).
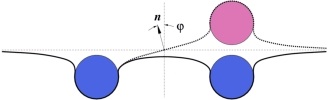 Soft interfaces can mediate interactions between particles bound to them. The force transmitted through the surface geometry on a particle may be expressed as a closed line integral of the surface stress tensor around that particle. This contour may be deformed to exploit the symmetries present; for two identical particles, one obtains an exact expression for the force between them in terms of the local surface geometry of their mid-plane; in the case of a fluid membrane the sign of the interaction is often evident. The approach, by construction, is adapted directly to the surface and is independent of its parameterization. Furthermore, it is applicable for arbitrarily large deformations; in particular, it remains valid beyond the linear small-gradient regime. Soft interfaces can mediate interactions between particles bound to them. The force transmitted through the surface geometry on a particle may be expressed as a closed line integral of the surface stress tensor around that particle. This contour may be deformed to exploit the symmetries present; for two identical particles, one obtains an exact expression for the force between them in terms of the local surface geometry of their mid-plane; in the case of a fluid membrane the sign of the interaction is often evident. The approach, by construction, is adapted directly to the surface and is independent of its parameterization. Furthermore, it is applicable for arbitrarily large deformations; in particular, it remains valid beyond the linear small-gradient regime.
|
|
2004 |
| |
When do fluid membranes engulf sticky colloids?
Markus Deserno
J. Phys.: Condens. Matter 16, S2061-S2070 (2004).
 Binding of a spherical colloid to a fluid membrane, which is an interplay between the energies of adhesion and elastic deformation, is studied within the framework of a Helfrich Hamiltonian. The solution of the full nonlinear shape equations for the membrane profile reveals a continuous binding and a discontinuous envelopment transition, the latter with a tension dependent substantial energy barrier. In the bending dominated regime this scenario is analytically confirmed by a small gradient expansion. Binding of a spherical colloid to a fluid membrane, which is an interplay between the energies of adhesion and elastic deformation, is studied within the framework of a Helfrich Hamiltonian. The solution of the full nonlinear shape equations for the membrane profile reveals a continuous binding and a discontinuous envelopment transition, the latter with a tension dependent substantial energy barrier. In the bending dominated regime this scenario is analytically confirmed by a small gradient expansion.
|
|
Screening of Spherical Colloids beyond Mean Field—A Local Density Functional Approach
Marcia C. Barbosa, Markus Deserno, Christian Holm, and René Messina
Phys. Rev. E 69, 051401 (2004).
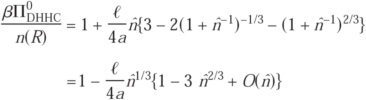 We study the counterion distribution around a spherical macroion and its osmotic pressure in the framework of the recently developed Debye-Hückel-Hole-Cavity (DHHC) theory. This is a local density functional approach which incorporates correlations into Poisson-Boltzmann theory by adding a free energy correction based on the One Component Plasma. We compare the predictions for ion distribution and osmotic pressure obtained by the full theory and by its zero temperature limit with Monte Carlo simulations. They agree excellently for weakly developed correlations and give the correct trend for stronger ones. In all investigated cases the DHHC theory and its computationally simpler zero temperature limit yield better results than the Poisson-Boltzmann theory. We study the counterion distribution around a spherical macroion and its osmotic pressure in the framework of the recently developed Debye-Hückel-Hole-Cavity (DHHC) theory. This is a local density functional approach which incorporates correlations into Poisson-Boltzmann theory by adding a free energy correction based on the One Component Plasma. We compare the predictions for ion distribution and osmotic pressure obtained by the full theory and by its zero temperature limit with Monte Carlo simulations. They agree excellently for weakly developed correlations and give the correct trend for stronger ones. In all investigated cases the DHHC theory and its computationally simpler zero temperature limit yield better results than the Poisson-Boltzmann theory.
|
|
Twist-bend instability for toroidal DNA condensates
Igor M. Kulic, Denis Andrienko, and Markus Deserno
Europhys. Lett. 67, 418–424 (2004).
 We propose that semiflexible polymers in poor solvent collapse in two stages. The first stage is the well known formation of a dense toroidal aggregate. However, if the solvent is sufficiently poor, the condensate will undergo a second structural transition to a twisted entangled state, in which individual filaments lower their bending energy by additionally orbiting around the mean path along which they wind. This ``topological ripening'' is consistent with known simulations and experimental results. It connects and rationalizes various experimental observations ranging from strong DNA entanglement in viral capsids to the unusually short pitch of the cholesteric phase of DNA in sperm-heads. We propose that topological ripening of DNA toroids could improve the efficiency and stability of gene delivery. We propose that semiflexible polymers in poor solvent collapse in two stages. The first stage is the well known formation of a dense toroidal aggregate. However, if the solvent is sufficiently poor, the condensate will undergo a second structural transition to a twisted entangled state, in which individual filaments lower their bending energy by additionally orbiting around the mean path along which they wind. This ``topological ripening'' is consistent with known simulations and experimental results. It connects and rationalizes various experimental observations ranging from strong DNA entanglement in viral capsids to the unusually short pitch of the cholesteric phase of DNA in sperm-heads. We propose that topological ripening of DNA toroids could improve the efficiency and stability of gene delivery.
|
|
A Statistical-Thermodynamic Model of Viral Budding
Shelly Tzlil, Markus Deserno, William M. Gelbart, and Avinoam Ben-Shaul
Biophys. J. 86, 2037–2048 (2004).
 We present a simple statistical thermodynamic model for budding of viral nucleocapsids at the cell membrane. The membrane is modeled as a flexible lipid bilayer embedding linker (``spike'') proteins, which serve to anchor and thus wrap the membrane around the viral capsids. The free energy of a single bud is expressed as a sum of the bending energy of its membrane coat, the spike-mediated capsid-membrane adhesion energy, and the line energy associated with the bud's rim; all depending on the extent of wrapping (i.e., bud size), and density of spikes in the curved membrane. This ``self-energy'' is incorporated into a simple free energy functional for the many-bud system, allowing for different spike densities, and hence entropy, in the curved (budding) and planar membrane regions, as well as for the configurational entropy of the polydisperse bud population. The equilibrium spike densities in the coexisting, curved and planar, membrane regions are calculated as a function of the membrane bending energy and the spike-mediated adhesion energy, for different spike and nucleocapsid concentrations in the membrane plane, as well as for several values of the bud's rim energy. We show that complete budding (full wrapping of nucleocapsids) can only take place if the adhesion energy exceeds a certain, critical, bending free energy. Whenever budding takes place, the spike density in the mature virions is saturated, i.e., all spike adhesion sites are occupied. The rim energy plays an important role in determining the size distribution of buds. The fraction of fully wrapped buds increases as this energy increases, resulting eventually in an ``all or nothing'' mechanism, whereby nucleocapsids at the plasma membrane are either fully enveloped or completely ``naked'' (just touching the membrane). We also find that at low concentrations all capsids arriving at the membrane get tightly and fully enveloped. Beyond a certain concentration, corresponding approximately to a ``stoichiometric'' spike-to-capsid ratio, newly arriving capsids cannot be fully wrapped, i.e., the budding yield decreases. We present a simple statistical thermodynamic model for budding of viral nucleocapsids at the cell membrane. The membrane is modeled as a flexible lipid bilayer embedding linker (``spike'') proteins, which serve to anchor and thus wrap the membrane around the viral capsids. The free energy of a single bud is expressed as a sum of the bending energy of its membrane coat, the spike-mediated capsid-membrane adhesion energy, and the line energy associated with the bud's rim; all depending on the extent of wrapping (i.e., bud size), and density of spikes in the curved membrane. This ``self-energy'' is incorporated into a simple free energy functional for the many-bud system, allowing for different spike densities, and hence entropy, in the curved (budding) and planar membrane regions, as well as for the configurational entropy of the polydisperse bud population. The equilibrium spike densities in the coexisting, curved and planar, membrane regions are calculated as a function of the membrane bending energy and the spike-mediated adhesion energy, for different spike and nucleocapsid concentrations in the membrane plane, as well as for several values of the bud's rim energy. We show that complete budding (full wrapping of nucleocapsids) can only take place if the adhesion energy exceeds a certain, critical, bending free energy. Whenever budding takes place, the spike density in the mature virions is saturated, i.e., all spike adhesion sites are occupied. The rim energy plays an important role in determining the size distribution of buds. The fraction of fully wrapped buds increases as this energy increases, resulting eventually in an ``all or nothing'' mechanism, whereby nucleocapsids at the plasma membrane are either fully enveloped or completely ``naked'' (just touching the membrane). We also find that at low concentrations all capsids arriving at the membrane get tightly and fully enveloped. Beyond a certain concentration, corresponding approximately to a ``stoichiometric'' spike-to-capsid ratio, newly arriving capsids cannot be fully wrapped, i.e., the budding yield decreases.
|
|
Elastic deformation of a fluid membrane upon colloid binding
Markus Deserno
Phys. Rev. E 69, 031903 (2004).
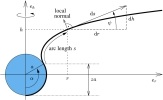 When a colloidal particle adheres to a fluid membrane, it induces elastic deformations in the membrane which oppose its own binding. The structural and energetic aspects of this balance are investigated within the framework of a Helfrich Hamiltonian. Based on the full nonlinear shape equations for the membrane profile, a line of continuous binding transitions and a second line of discontinuous envelopment transitions are found, which meet at an unusual triple point. The regime of low tension is studied analytically using a small gradient expansion, while in the limit of large tension scaling arguments are derived which quantify the asymptotic behavior of phase boundary, degree of wrapping, and energy barrier. The maturation of animal viruses by budding is discussed as a biological example of such colloid-membrane interaction events. When a colloidal particle adheres to a fluid membrane, it induces elastic deformations in the membrane which oppose its own binding. The structural and energetic aspects of this balance are investigated within the framework of a Helfrich Hamiltonian. Based on the full nonlinear shape equations for the membrane profile, a line of continuous binding transitions and a second line of discontinuous envelopment transitions are found, which meet at an unusual triple point. The regime of low tension is studied analytically using a small gradient expansion, while in the limit of large tension scaling arguments are derived which quantify the asymptotic behavior of phase boundary, degree of wrapping, and energy barrier. The maturation of animal viruses by budding is discussed as a biological example of such colloid-membrane interaction events.
|
|
2003 |
| |
Osmotic Shock and the Strength of Viral Capsids
Amado Cordova, Markus Deserno, William M. Gelbart, and Avinoam Ben-Shaul
Biophys. J. 85, 70–74 (2003).
 Osmotic shock is a familiar means for rupturing viral capsids and exposing their genomes intact. The necessary conditions for providing this shock involve incubation in high-concentration salt solutions, and lower permeability of the capsids to salt ions than to water molecules. We discuss here how values of the capsid strength can be inferred from calculations of the osmotic pressure differences associated with measured values of the critical concentration of incubation solution. Osmotic shock is a familiar means for rupturing viral capsids and exposing their genomes intact. The necessary conditions for providing this shock involve incubation in high-concentration salt solutions, and lower permeability of the capsids to salt ions than to water molecules. We discuss here how values of the capsid strength can be inferred from calculations of the osmotic pressure differences associated with measured values of the critical concentration of incubation solution.
|
|
Wrapping of a spherical colloid by a fluid membrane
Markus Deserno and Thomas Bickel
Europhys. Lett. 62, 767–773 (2003).
 We theoretically study the elastic deformation of a fluid membrane induced by an adhering spherical colloidal particle within the framework of a Helfrich energy. Based on a full optimization of the membrane shape we find a continuous binding and a discontinuous envelopment transition, the latter displaying a potentially substantial energy barrier. A small-gradient approximation permits membrane shape and complex energy to be calculated analytically. While this only leads to a good representation of the complex geometry for very small degrees of wrapping, it still gives the correct phase boundaries in the regime of low tension. We theoretically study the elastic deformation of a fluid membrane induced by an adhering spherical colloidal particle within the framework of a Helfrich energy. Based on a full optimization of the membrane shape we find a continuous binding and a discontinuous envelopment transition, the latter displaying a potentially substantial energy barrier. A small-gradient approximation permits membrane shape and complex energy to be calculated analytically. While this only leads to a good representation of the complex geometry for very small degrees of wrapping, it still gives the correct phase boundaries in the regime of low tension.
|
|
Attraction and ionic correlations between charged stiff polyelectrolytes
Markus Deserno, Axel Arnold, and Christian Holm
Macromolecules 36, 249–259 (2003).
 We use molecular dynamics simulations to study attractive interactions and the underlying ionic correlations between parallel like-charged rods in the absence of additional salt. For a generic bulk system of rods we identify a reduction of short-range repulsions as the origin of a negative osmotic coefficient. The counterions show signs of a weak three-dimensional order in the attractive regime only once the rod-imposed charge inhomogeneities are divided out. We also treat the case of attraction between a single pair of rods for a few selected line charge densities and rod radii. Measurements of the individual contributions to the force between close rods are studied as a function of Bjerrum length. We find that even though the total force is always attractive at sufficiently high Bjerrum length, the electrostatic contribution can ultimately become repulsive. We also measure azimuthal and longitudinal correlation functions to answer the question how condensed ions are distributed with respect to each other and to the neighboring rod. For instance, we show that the prevalent image of mutually interlocked ions is qualitatively correct, even though modifications due to thermal fluctuations are usually strong. We use molecular dynamics simulations to study attractive interactions and the underlying ionic correlations between parallel like-charged rods in the absence of additional salt. For a generic bulk system of rods we identify a reduction of short-range repulsions as the origin of a negative osmotic coefficient. The counterions show signs of a weak three-dimensional order in the attractive regime only once the rod-imposed charge inhomogeneities are divided out. We also treat the case of attraction between a single pair of rods for a few selected line charge densities and rod radii. Measurements of the individual contributions to the force between close rods are studied as a function of Bjerrum length. We find that even though the total force is always attractive at sufficiently high Bjerrum length, the electrostatic contribution can ultimately become repulsive. We also measure azimuthal and longitudinal correlation functions to answer the question how condensed ions are distributed with respect to each other and to the neighboring rod. For instance, we show that the prevalent image of mutually interlocked ions is qualitatively correct, even though modifications due to thermal fluctuations are usually strong.
|
|
2002 |
| |
Theory and simulations of rigid polyelectrolytes
Markus Deserno and Christian Holm
Mol. Phys. 100, 2941–2956 (2002).
 Theoretical and numerical studies are reported on stiff, linear polyelectrolytes within the framework of the cell model, first reviewing analytical results obtained on a mean-field Poisson-Boltzmann level, and then using molecular dynamics simulations to show the circumstances under which these fail quantitatively and qualitatively. For the hexagonally packed nematic phase of the polyelectrolytes the osmotic coefficient is computed as a function of density. In the presence of multivalent counterions it can become negative, leading to effective attractions. This is shown to result from a reduced contribution of the virial part to the pressure. The osmotic coefficient and ionic distribution functions are computed from Poisson-Boltzmann theory with and without a recently proposed correlation correction. Simulation results for the case of poly(p-phenylene) are presented and compared with recently obtained experimental data on this stiff polyelectrolyte. Ion-ion correlations in the strong coupling regime are studied and compared with the predictions of the recently advocated Wigner crystal theories. Theoretical and numerical studies are reported on stiff, linear polyelectrolytes within the framework of the cell model, first reviewing analytical results obtained on a mean-field Poisson-Boltzmann level, and then using molecular dynamics simulations to show the circumstances under which these fail quantitatively and qualitatively. For the hexagonally packed nematic phase of the polyelectrolytes the osmotic coefficient is computed as a function of density. In the presence of multivalent counterions it can become negative, leading to effective attractions. This is shown to result from a reduced contribution of the virial part to the pressure. The osmotic coefficient and ionic distribution functions are computed from Poisson-Boltzmann theory with and without a recently proposed correlation correction. Simulation results for the case of poly(p-phenylene) are presented and compared with recently obtained experimental data on this stiff polyelectrolyte. Ion-ion correlations in the strong coupling regime are studied and compared with the predictions of the recently advocated Wigner crystal theories.
|
|
The osmotic pressure of charged colloidal suspensions: A unified approach to linearized Poisson-Boltzmann theory
Markus Deserno and Hans-Hennig von Grünberg
Phys. Rev. E 66, 011401 (2002).
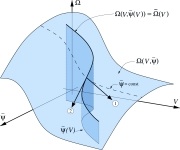 We study theoretically the equation of state of a fluid suspension of charged objects (e.g., colloids, polyelectrolytes, clay platelets, etc.) dialyzed against an electrolyte solution using the cell model and linear Poisson-Boltzmann (PB) theory. From the volume derivative of the grand potential functional of linear theory we obtain two expressions for the osmotic pressure in terms of the potential or ion profiles, neither of which coincides with the expression known from nonlinear PB theory, namely, the density of microions at the cell boundary. We show that the range of validity of linearization depends strongly on the linearization point and prove that expansion about the self-consistently determined average potential is optimal in several respects. For instance, screening inside the suspension is automatically described by the actual ionic strength, resulting in the correct asymptotics at high colloid concentration. Together with the analytical solution of the linear PB equation for cell models of arbitrary dimension and electrolyte composition, explicit and very general formulas for the osmotic pressure ensue. A comparison with nonlinear PB theory is provided. Our analysis also shows that whether or not linear theory predicts a phase separation depends crucially on the precise definition of the pressure, showing that depending on the choice, an artificial phase separation in systems as important as DNA in physiological salt solution may result. We study theoretically the equation of state of a fluid suspension of charged objects (e.g., colloids, polyelectrolytes, clay platelets, etc.) dialyzed against an electrolyte solution using the cell model and linear Poisson-Boltzmann (PB) theory. From the volume derivative of the grand potential functional of linear theory we obtain two expressions for the osmotic pressure in terms of the potential or ion profiles, neither of which coincides with the expression known from nonlinear PB theory, namely, the density of microions at the cell boundary. We show that the range of validity of linearization depends strongly on the linearization point and prove that expansion about the self-consistently determined average potential is optimal in several respects. For instance, screening inside the suspension is automatically described by the actual ionic strength, resulting in the correct asymptotics at high colloid concentration. Together with the analytical solution of the linear PB equation for cell models of arbitrary dimension and electrolyte composition, explicit and very general formulas for the osmotic pressure ensue. A comparison with nonlinear PB theory is provided. Our analysis also shows that whether or not linear theory predicts a phase separation depends crucially on the precise definition of the pressure, showing that depending on the choice, an artificial phase separation in systems as important as DNA in physiological salt solution may result.
|
|
Adhesion and Wrapping in Colloid-Vesicle Complexes
Markus Deserno and William M. Gelbart
J. Phys. Chem. B 106, 5543–5552 (2002).
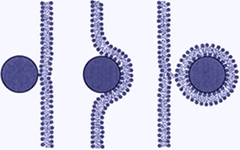 We present a simple theoretical model for adsorption of colloidal particles onto vesicles. The contact energy of adhesion is balanced by the tension and curvature energies of the vesicle membrane under the constraint of fixed volume, with the geometry of the complex determined by a variational calculation. Physical observables, such as the degree of penetration or the membrane tension, are investigated as functions of colloidal size and adhesion, tension, and bending energies. We find various new (and discontinuous) transitions in the geometry of the complex compared to those from a description that neglects the curvature contribution. Particular emphasis is put on the transition from the partly to the fully wrapped state and on unbinding of the complex at weak adhesion energy or small colloidal size. The above model can be thought of as a phenomenological theory of the initial steps involved in biological endocytosis and aims toward an improved physical understanding of this process. We present a simple theoretical model for adsorption of colloidal particles onto vesicles. The contact energy of adhesion is balanced by the tension and curvature energies of the vesicle membrane under the constraint of fixed volume, with the geometry of the complex determined by a variational calculation. Physical observables, such as the degree of penetration or the membrane tension, are investigated as functions of colloidal size and adhesion, tension, and bending energies. We find various new (and discontinuous) transitions in the geometry of the complex compared to those from a description that neglects the curvature contribution. Particular emphasis is put on the transition from the partly to the fully wrapped state and on unbinding of the complex at weak adhesion energy or small colloidal size. The above model can be thought of as a phenomenological theory of the initial steps involved in biological endocytosis and aims toward an improved physical understanding of this process.
|
|
2001 |
| |
Rayleigh instability of charged droplets in the presence of counterions
Markus Deserno
Eur. Phys. J. E 6, 163–168 (2001).
 The classical instability of a charged spherical droplet is reconsidered in the presence of counterions. An ensemble of such droplets is studied within a simplified cell model. Screening of the electric field by the counterions is found to increase the equilibrium droplet size. Furthermore, if the ions can enter the droplet, a first-order phase transition occurs upon increasing Bjerrum length, surface tension or droplet density, leading to a phase separation. Simple scaling properties of the free energy give the shape of the phase boundary and show the system to be scale-invariant there. Pearl-necklace structures of hydrophobic polyelectrolytes are discussed as an application. The classical instability of a charged spherical droplet is reconsidered in the presence of counterions. An ensemble of such droplets is studied within a simplified cell model. Screening of the electric field by the counterions is found to increase the equilibrium droplet size. Furthermore, if the ions can enter the droplet, a first-order phase transition occurs upon increasing Bjerrum length, surface tension or droplet density, leading to a phase separation. Simple scaling properties of the free energy give the shape of the phase boundary and show the system to be scale-invariant there. Pearl-necklace structures of hydrophobic polyelectrolytes are discussed as an application.
|
|
Overcharging of DNA in the Presence of Salt: Theory and Simulation
Markus Deserno, Felipe Jiménez-Ángeles, Christian Holm, and Marcelo Lozada-Cassou
J. Phys. Chem. B 105, 10983–10991 (2001).
 A study of a model rodlike polyelectrolyte molecule immersed in a monovalent or divalent electrolyte is presented. Results for the local concentration profile, mean electrostatic potential, charge distribution function, and zeta-potential are obtained from hypernetted-chain/mean spherical approximation (HNC/MSA) theory and compared with molecular dynamics (MD) simulations. As a particular case, the parameters of the polyelectrolyte molecule are mapped to those of a DNA molecule. Both HNC/MSA and MD predict the occurrence of overcharging, which is not present in the Poisson-Boltzmann theory. Further an excellent qualitative, and in some cases quantitative, agreement between HNC/MSA and MD is found. Oscillations observed in the mean electrostatic potential, local concentration profiles, and the curvature of the zeta-potential are discussed in terms of the observed overcharging effect. Particularly interesting results are a very nonmonotonic behavior of the zeta-potential, as a function of the rod charge density, and the overcharging by monovalent counterions. A study of a model rodlike polyelectrolyte molecule immersed in a monovalent or divalent electrolyte is presented. Results for the local concentration profile, mean electrostatic potential, charge distribution function, and zeta-potential are obtained from hypernetted-chain/mean spherical approximation (HNC/MSA) theory and compared with molecular dynamics (MD) simulations. As a particular case, the parameters of the polyelectrolyte molecule are mapped to those of a DNA molecule. Both HNC/MSA and MD predict the occurrence of overcharging, which is not present in the Poisson-Boltzmann theory. Further an excellent qualitative, and in some cases quantitative, agreement between HNC/MSA and MD is found. Oscillations observed in the mean electrostatic potential, local concentration profiles, and the curvature of the zeta-potential are discussed in terms of the observed overcharging effect. Particularly interesting results are a very nonmonotonic behavior of the zeta-potential, as a function of the rod charge density, and the overcharging by monovalent counterions.
|
|
The osmotic coefficient of rod-like polyelectrolytes: Computer simulation, analytical theory, and experiment
Markus Deserno, Christian Holm, Jürgen Blaul, Matthias Ballauff, and Matthias Rehahn
Eur. Phys. J. E 5, 97–103 (2001).
 The osmotic coefficient of solutions of rod-like polyelectrolytes is considered by comparing current theoretical treatments and simulations to recent experimental data. The discussion is restricted to the case of monovalent counterions and dilute, salt-free solutions. The classical Poisson-Boltzmann solution of the cell model correctly predicts a strong decrease in the osmotic coefficient, but upon closer look systematically overestimates its value. The contribution of ion-ion-correlations are quantitatively studied by MD simulations and the recently proposed DHHC theory. However, our comparison with experimental data obtained on synthetic, stiff-chain polyelectrolytes shows that correlation effects can only partly explain the discrepancy. A quantitative understanding thus requires theoretical efforts beyond the restricted primitive model of electrolytes. The osmotic coefficient of solutions of rod-like polyelectrolytes is considered by comparing current theoretical treatments and simulations to recent experimental data. The discussion is restricted to the case of monovalent counterions and dilute, salt-free solutions. The classical Poisson-Boltzmann solution of the cell model correctly predicts a strong decrease in the osmotic coefficient, but upon closer look systematically overestimates its value. The contribution of ion-ion-correlations are quantitatively studied by MD simulations and the recently proposed DHHC theory. However, our comparison with experimental data obtained on synthetic, stiff-chain polyelectrolytes shows that correlation effects can only partly explain the discrepancy. A quantitative understanding thus requires theoretical efforts beyond the restricted primitive model of electrolytes.
|
|
2000 |
| |
Ion-ion correlations: an improved one-component plasma correction
Marcia C. Barbosa, Markus Deserno, and Christian Holm
Europhys. Lett. 52, 80–86 (2000).
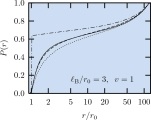 We propose a new approach for incorporating counterion correlations into Poisson-Boltzmann-like theories, which is based on a local density functional for the system of counterions. Our derivation is inspired by the Debye-Hückel approach to the one-component plasma (OCP). An artificial instability due to the OCP's homogeneous background charge – which is absent in the original counterion system – is overcome by excluding the background from the vicinity of a central ion. The resulting free energy density is convex and thus applicable within a local density approximation. For the simple example of a strongly charged stiff rod surrounded by its counterions we demonstrate that the Poisson-Boltzmann free energy functional augmented by our new correction accounts for the correlations present in this system when compared to molecular dynamics simulations. We propose a new approach for incorporating counterion correlations into Poisson-Boltzmann-like theories, which is based on a local density functional for the system of counterions. Our derivation is inspired by the Debye-Hückel approach to the one-component plasma (OCP). An artificial instability due to the OCP's homogeneous background charge – which is absent in the original counterion system – is overcome by excluding the background from the vicinity of a central ion. The resulting free energy density is convex and thus applicable within a local density approximation. For the simple example of a strongly charged stiff rod surrounded by its counterions we demonstrate that the Poisson-Boltzmann free energy functional augmented by our new correction accounts for the correlations present in this system when compared to molecular dynamics simulations.
|
|
A Monte-Carlo approach to Poisson-Boltzmann like free-energy functionals
Markus Deserno
Physica A 278, 405–413 (2000).
 A simple technique is proposed for numerically determining equilibrium ion distribution functions belonging to free energies of the Poisson-Boltzmann type. The central idea is to perform a conventional Monte-Carlo simulation using the free energy as the "Hamiltonian" entering the Metropolis criterion and the spatially discretized density as degrees of freedom. This approach is complementary to the possibility of numerically solving the differential equations corresponding to the variational problem, but it is much easier to implement and to generalize. Its utility is demonstrated in two examples: valence mixtures and hard core interactions of ions surrounding a charged rod. A simple technique is proposed for numerically determining equilibrium ion distribution functions belonging to free energies of the Poisson-Boltzmann type. The central idea is to perform a conventional Monte-Carlo simulation using the free energy as the "Hamiltonian" entering the Metropolis criterion and the spatially discretized density as degrees of freedom. This approach is complementary to the possibility of numerically solving the differential equations corresponding to the variational problem, but it is much easier to implement and to generalize. Its utility is demonstrated in two examples: valence mixtures and hard core interactions of ions surrounding a charged rod.
|
|
Fraction of condensed counterions around a charged rod: Comparison of Poisson-Boltzmann theory and computer simulations
Markus Deserno, Christian Holm, and Sylvio May
Macromolecules 33, 199–206 (2000).
 We investigate the phenomenon of counterion condensation in a solution of highly charged rigid polyelectrolytes within the cell model. A method is proposed which-based on the charge distribution function-identifies both the fraction of condensed ions and the radial extension of the condensed layer. Within salt-free Poisson-Boltzmann (PB) theory it reproduces the well-known fraction 1-1/ξ of condensed ions for a Manning parameter ξ > 1. Furthermore, it predicts a weak salt dependence of this fraction and a breakdown of the concept of counterion condensation in the high-salt limit. We complement our theoretical investigations with molecular dynamics simulations of a cell-like model, which constantly yield a stronger condensation than predicted by PB theory. While the agreement between theory and simulation is excellent in the monovalent, weakly charged case, it deteriorates with increasing electrostatic interaction strength and, in particular, increasing valence. For instance, at a high concentration of divalent salt and large xi our computer simulations predict charge oscillations, which mean-field theory is unable to reproduce. We investigate the phenomenon of counterion condensation in a solution of highly charged rigid polyelectrolytes within the cell model. A method is proposed which-based on the charge distribution function-identifies both the fraction of condensed ions and the radial extension of the condensed layer. Within salt-free Poisson-Boltzmann (PB) theory it reproduces the well-known fraction 1-1/ξ of condensed ions for a Manning parameter ξ > 1. Furthermore, it predicts a weak salt dependence of this fraction and a breakdown of the concept of counterion condensation in the high-salt limit. We complement our theoretical investigations with molecular dynamics simulations of a cell-like model, which constantly yield a stronger condensation than predicted by PB theory. While the agreement between theory and simulation is excellent in the monovalent, weakly charged case, it deteriorates with increasing electrostatic interaction strength and, in particular, increasing valence. For instance, at a high concentration of divalent salt and large xi our computer simulations predict charge oscillations, which mean-field theory is unable to reproduce.
|
|
1998 |
| |
How to mesh up Ewald sums (II): An accurate error estimate for the P3M algorithm
Markus Deserno and Christian Holm
J. Chem. Phys. 109, 7694–7701 (1998).
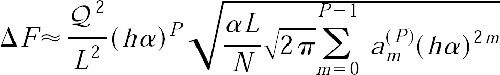 We construct an accurate estimate for the root mean square force error of the particle-particle–particle-mesh (P3M) algorithm by extending a single particle pair error measure which has been given by Hockney and Eastwood. We also derive an easy-to-use analytic approximation to the error formula. This allows a straightforward and precise determination of the optimal splitting parameter (as a function of system specifications and P3M parameters) and hence knowledge of the force accuracy prior to the actual simulation. The high quality of the estimate is demonstrated in several examples. We construct an accurate estimate for the root mean square force error of the particle-particle–particle-mesh (P3M) algorithm by extending a single particle pair error measure which has been given by Hockney and Eastwood. We also derive an easy-to-use analytic approximation to the error formula. This allows a straightforward and precise determination of the optimal splitting parameter (as a function of system specifications and P3M parameters) and hence knowledge of the force accuracy prior to the actual simulation. The high quality of the estimate is demonstrated in several examples.
|
|
How to mesh up Ewald sums (I): A theoretical and numerical comparison of various particle mesh routines
Markus Deserno and Christian Holm
J. Chem. Phys. 109, 7678–7693 (1998).
 Standard Ewald sums, which calculate e.g. the electrostatic energy or the force in periodically closed systems of charged particles, can be efficiently speeded up by the use of the Fast Fourier Transformation (FFT). In this article we investigate three algorithms for the FFT-accelerated Ewald sum, which attracted a widespread attention, namely, the so-called particle-particle–particle-mesh (P3M), particle mesh Ewald (PME) and smooth PME method. We present a unified view of the underlying techniques and the various ingredients which comprise those routines. Additionally, we offer detailed accuracy measurements, which shed some light on the influence of several tuning parameters and also show that the existing methods – although similar in spirit – exhibit remarkable differences in accuracy. We propose combinations of the individual components, mostly relying on the P3M approach, which we regard as most flexible. The issue of estimating the errors connected with particle mesh routines is reserved to part II of this series. Standard Ewald sums, which calculate e.g. the electrostatic energy or the force in periodically closed systems of charged particles, can be efficiently speeded up by the use of the Fast Fourier Transformation (FFT). In this article we investigate three algorithms for the FFT-accelerated Ewald sum, which attracted a widespread attention, namely, the so-called particle-particle–particle-mesh (P3M), particle mesh Ewald (PME) and smooth PME method. We present a unified view of the underlying techniques and the various ingredients which comprise those routines. Additionally, we offer detailed accuracy measurements, which shed some light on the influence of several tuning parameters and also show that the existing methods – although similar in spirit – exhibit remarkable differences in accuracy. We propose combinations of the individual components, mostly relying on the P3M approach, which we regard as most flexible. The issue of estimating the errors connected with particle mesh routines is reserved to part II of this series.
|
|
1997 |
| |
Tricriticality and the Blume-Capel model: A Monte Carlo study within the microcanonical ensemble
Markus Deserno
Phys. Rev. E 56, 5204–5210 (1997).
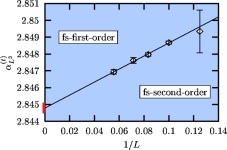 The microcanonical partition function Ωα,N(E) of a three-dimensional ferromagnetic simple cubic Blume-Capel model is calculated for several coupling ratios alpha close to and on the first-order side of the tricritical point. To this end a single Monte Carlo simulation of a suitably extended partition function for systems of L×L×L spins with L in {8,10,12,14,18} was performed. A finite system analog of the latent heat is introduced in order to define a tricritical point in finite systems as well. An empirical scaling of its coordinates to the thermodynamic limit yields αt=2.844 79 +- 0.00030 for the tricritical coupling ratio, and kTt/J=1.4182 +- 0.0055 for the tricritical transition temperature. The results are compared with values obtained on FCC lattices. The microcanonical partition function Ωα,N(E) of a three-dimensional ferromagnetic simple cubic Blume-Capel model is calculated for several coupling ratios alpha close to and on the first-order side of the tricritical point. To this end a single Monte Carlo simulation of a suitably extended partition function for systems of L×L×L spins with L in {8,10,12,14,18} was performed. A finite system analog of the latent heat is introduced in order to define a tricritical point in finite systems as well. An empirical scaling of its coordinates to the thermodynamic limit yields αt=2.844 79 +- 0.00030 for the tricritical coupling ratio, and kTt/J=1.4182 +- 0.0055 for the tricritical transition temperature. The results are compared with values obtained on FCC lattices.
|
|
| |
Proceeding, Schools, ... |
| |
Lipid Membranes: From Self-assembly to Elasticity
M. Mert Terzi and Markus Deserno
in: The Role of Mechanics in the Study of Lipid Bilayers, CISM International Centre for Mechanical Sciences Courses and Lectures Volume 577, edited by David Steigmann; Springer Nature (Cham, Switzerland, 2018).
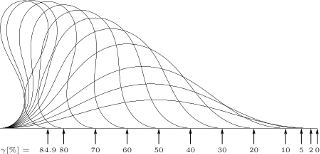 In aqueous solution, lipid molecules spontaneously assemble into macro- scopic bilayer membranes, which have highly interesting mechanical properties. In this chapter, we first discuss some basic aspects of this self-assembly process. In the second part, we then revisit and slightly expand a well-known continuum-level the- ory that describes the elastic properties pertaining to membrane geometry and lipid tilt. We then illustrate in part three several conceptually different strategies for how one of the emerging elastic parameters—the bending modulus—can be obtained in computer simulations. In aqueous solution, lipid molecules spontaneously assemble into macro- scopic bilayer membranes, which have highly interesting mechanical properties. In this chapter, we first discuss some basic aspects of this self-assembly process. In the second part, we then revisit and slightly expand a well-known continuum-level the- ory that describes the elastic properties pertaining to membrane geometry and lipid tilt. We then illustrate in part three several conceptually different strategies for how one of the emerging elastic parameters—the bending modulus—can be obtained in computer simulations.
|
|
Membrane Elasticity and Mediated Interactions in Continuum Theory: A Differential Geometric Approach
Markus Deserno
in: Biomembrane Frontiers: Nanostructures, Models, and the Design of Life, Volume 2, edited by Roland Faller, Thomas Jue, Marjorie L. Longo, and Subhash H. Risbud,Humana Press (Springer, New York, 2009).
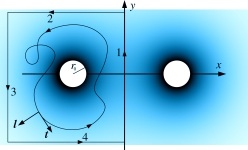 Objects in contact with membranes can locally deform these membranes, and several such objects can interact by means of the elastic stresses thus created. For fluid surfaces the Hamiltonian is purely geometric, and this permits a very efficient treatment of this scenario using methods of covariant differential geometry. This article provides a pedagogical introduction into the philosophy behind this approach. The relation between geometry and elasticity is first derived in a very intuitive way and then extended to more complex (but very relevant) examples. The relation between stresses and geometry is introduced and illustrated in three very different examples: interactions mediated by (a) composition variations, (b) lipid tilt fields, and (c) membrane curvature. Objects in contact with membranes can locally deform these membranes, and several such objects can interact by means of the elastic stresses thus created. For fluid surfaces the Hamiltonian is purely geometric, and this permits a very efficient treatment of this scenario using methods of covariant differential geometry. This article provides a pedagogical introduction into the philosophy behind this approach. The relation between geometry and elasticity is first derived in a very intuitive way and then extended to more complex (but very relevant) examples. The relation between stresses and geometry is introduced and illustrated in three very different examples: interactions mediated by (a) composition variations, (b) lipid tilt fields, and (c) membrane curvature.
|
|
Solvent-free lipid-bilayer simulations: From Physics to Biology
Markus Deserno
in: Computer Simulation Studies in Condensed Matter Physics XIX, Eds. D.P. Landau, S.P. Lewis, and H.B. Schüttler (Springer, Heidelberg, Berlin, 2006).
Coarse-grained solvent-free simulation models enabling the study of self-assembling fluid lipid bilayers have been the goal of much recent modeling efforts, since their realization appeared to be quite intricate. This contribution reviews some of the challenges faced along the way, presents a surprisingly simple solution, and illustrates its capacity by means of three examples of biological interest. |
|
Cell Model and Poisson-Boltzmann Theory: A brief introduction
Markus Deserno and Christian Holm
in: Electrostatic Effects in Soft Matter and Biophysics, Vol 46 of NATO Advanced Studies Institute, Series II: Mathematics, Physics and Chemistry, ed. by Christian Holm, Patrick Kékicheff, and Rudolf Podgornik (Kluwer, Dordrecht, 2001).
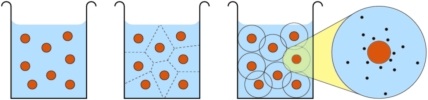 The cell-model and its treatment on the Poisson-Boltzmann level are two important concepts in the theoretical description of charged macromolecules. In this brief contribution we provide an introduction to both ideas and summarize a few important results which can be obtained from them. Our article is organized as follows: Section 1 outlines the sequence of approximations which ultimately lead to the cell-model. Section 2 is devoted to two exact results, namely, an expression for the osmotic pressure and a formula for the ion density at the surface of the macromolecule, known as the contact value theorem. Section 3 provides a derivation of the Poisson-Boltzmann equation from a variational principle and the assumption of a product state. Section 4 applies Poisson-Boltzmann theory to the cell-model of linear polyelectrolytes. In particular, the behavior of the exact solution in the limit of zero density is compared to the concept of Manning condensation. Finally, Section 5 shows how a system described by a cell model can be coupled to a salt reservoir, i.e., how the so-called Donnan equilibrium is established. The cell-model and its treatment on the Poisson-Boltzmann level are two important concepts in the theoretical description of charged macromolecules. In this brief contribution we provide an introduction to both ideas and summarize a few important results which can be obtained from them. Our article is organized as follows: Section 1 outlines the sequence of approximations which ultimately lead to the cell-model. Section 2 is devoted to two exact results, namely, an expression for the osmotic pressure and a formula for the ion density at the surface of the macromolecule, known as the contact value theorem. Section 3 provides a derivation of the Poisson-Boltzmann equation from a variational principle and the assumption of a product state. Section 4 applies Poisson-Boltzmann theory to the cell-model of linear polyelectrolytes. In particular, the behavior of the exact solution in the limit of zero density is compared to the concept of Manning condensation. Finally, Section 5 shows how a system described by a cell model can be coupled to a salt reservoir, i.e., how the so-called Donnan equilibrium is established.
|
|
Molecular Dynamics Simulation of the Cell Model
Markus Deserno, Christian Holm, and Kurt Kremer
in: Physical Chemistry of Polyelectrolytes, Vol. 99 of the surfactant science series, ed. by Tsetska Radeva (Marcel Dekker, New York, 2001).
|
| |
| |
| |
| |
| |
| |
| |
| |
| |
| |
| |
| |
| |
| |
|
|
|


